Biology Worthy of Life
An experiment in revivifying biology
How the Organism Decides What to Make of Its Genes
With Special Emphasis on the Human Being
Compiled by Stephen L. Talbott (stevet@netfuture.org)
RSS feed for the Biology Worthy of Life project:

Copyright 2013 – 2024 The Nature Institute. All rights reserved.
These are raw notes from my own reading of the literature of gene regulation. Please see the essential caveats in order to understand their limitations. Despite those limitations, the notes are presented here because browsing them should convince any reader — including those molecular biologists whose reading is largely confined to their own specialties — that it is the organism that makes use of its genes, not the other way around.
I periodically, if somewhat erratically, add further notes to this document, but am far less good at going through and deleting any outdated material, much less cleaning up disorganized aspects of the presentation. Nevertheless, I will welcome general comments and also suggestions for improving things. Send them to stevet@netfuture.org.
Please do read the brief introduction, which offers a useful perspective on these notes.
For most people, the best way to digest this document would not be to read straight through it from beginning to end, but merely to page through it, reading a few notes here and there in order to get a feel for the variety of processes at work in shaping how the organism makes use of its genes.
To get a sense for the scope of the overall document, you can:
- expand and collapse the submenus for specific entries in the table of contents by clicking on the "(+/-)" symbol after those entries
- Expand all menus
- Collapse all menus
Contents INTRODUCTION NEGOTIATIONS AMONG PARENTS AND OFFSPRING (+/-)
INTRODUCTION
“The remarkable complexity of gene regulation becomes increasingly apparent in proportion to the improving resolution of the available assays” (Kalsotra and Cooper 2011).
“Rather than thinking of gene regulation as an on/off process, we now have to accept that our genomes are pervasively transcribed; that regulatory noncoding RNAs (ncRNAs) ... complement transcription factors in gene regulation; that genes that are not expressed (which in the conventional sense are ‘off’) are often associated with engaged RNA polymerase II, producing short noncoding transcripts at their promoters; and that genes are differentially marked to respond to a particular developmental program long before they are actually expressed. Transcription factors are now associated with terms such as ‘rheostat’ rather than ‘switch,’ and, together with large coactivator protein complexes, often control the transition to the elongation stage of transcription. Not surprisingly, the chromatin environment in which genes reside has yielded some surprises, not least that there are new and uncharacterized chromatin domains, and challenges to the idea that histone modifications reflect the functionality of the complexes that deposit them” (Mellor 2010).
But all this only vaguely suggests a very few of the overwhelming variety of factors that influence how the organism makes use of its genes.
 “Understanding gene regulation is a central goal of molecular biology, which
remains only partially solved as DNA sequence alone cannot explain cell-type-
or context-specific gene activity. Instead, the coordinated activities of
thousands of regulatory proteins ensure expression of the correct set of
cell-type-specific proteins and enable rapid adaptation to changing
conditions”
(Goronzy 2025, doi:10.1038/s41580-025-00830-7).
“Understanding gene regulation is a central goal of molecular biology, which
remains only partially solved as DNA sequence alone cannot explain cell-type-
or context-specific gene activity. Instead, the coordinated activities of
thousands of regulatory proteins ensure expression of the correct set of
cell-type-specific proteins and enable rapid adaptation to changing
conditions”
(Goronzy 2025, doi:10.1038/s41580-025-00830-7).
NEGOTIATIONS AMONG PARENTS AND OFFSPRING
-
Maternal RNA
The earlier we look in the development of an organism, the more crucial
and far-reaching are the consequences of developmental activities. And
at the earliest stage of all — in the post-fertilization zygote — the
mRNA that is involved in protein generation, and the microRNAs and
other small RNAs that help regulate protein expression, are derived
from the mother (and to some degree from the father). The new
organism’s own genes are not yet active. It would be hard to overstate the
importance of this feature of whole-organism inheritance. (See
“Genes and the Central Fallacy of
Evolutionary Theory”.) This section, which is now nearly empty,
deserves massive treatment.
- Activation of maternal mRNA is regulated in the zygote, a regulation that, at least in zebrafish and mice (and presumably in other mammals), is achieved by polyadenylation of the mRNA, rendering it suitable for translation. Some of the mRNAs are activated in this way prior to fertilization, playing a role in early cleavage and the blastula; others are activated at around the time when the zygote’s own transcriptional processes are ready to begin (mid-blastula transition). Regulation also entails the timed degradation of maternal mRNAs, carried out by maternal microRNAs and probably also by other means (Aanes, Winata, Lin et al. 2011).
- “A fundamental principle in biology is that the program for early development is established during oogenesis in the form of the maternal transcriptome ... Here we show that 3' terminal uridylation of mRNA mediated by [the proteins] TUT4 and TUT7 sculpts the mouse maternal transcriptome by eliminating transcripts during oocyte growth. Uridylation mediated by TUT4 and TUT7 is essential for both oocyte maturation and fertility. In comparison to somatic cells, the oocyte transcriptome has a shorter poly(A) tail and a higher relative proportion of terminal oligo-uridylation. Deletion of TUT4 and TUT7 leads to the accumulation of a cohort of transcripts with a high frequency of very short poly(A) tails, and a loss of 3' oligo-uridylation. By contrast, deficiency of TUT4 and TUT7 does not alter gene expression in a variety of somatic cells. In summary, we show that poly(A) tail length and 3' terminal uridylation have essential and specific functions in shaping a functional maternal transcriptome” (Morgan, Much, DiGiacomo et al. 2017a, doi:10.1038/nature23318).
-
Paternal RNA
More recently it’s been demonstrated that some paternal mRNA makes its way
into the zygote. The implications of this are only now beginning to be
elucidated (Boerke and Gadella 2007; Dadoune 2009; Lalancette, Platts,
Johnson et al. 2009; Miller 2011).
“Increasing attention has focused on the significance of RNA in sperm, in
light of its contribution to the birth and long-term health of a child,
role in sperm function and diagnostic potential”. Examination of RNA in
sperm reveals “unique features indicative of very specific
and stage-dependent maturation and regulation of sperm RNA, illuminating
their various transitional roles. Correlation of sperm transcript abundance
with epigenetic marks suggested roles for these elements in the pre- and
post-fertilization genome. Several classes of non-coding RNAs including
long noncoding RNAs, chromatin-associated RNAs, pri-miRNAs [primary
microRNAs], novel elements and mRNAs have been identified which, based on
factors including relative abundance, integrity in sperm, available
knockout data of embryonic effect and presence or absence in the
unfertilized human oocyte, are likely to be essential male factors critical
to early post-fertilization development” (Sendler, Johnson, Mao et al.
2013).
- According to a story in Nature, research on mice shows that stress early in life can alter microRNAs in sperm, and these microRNAs can play a rather dramatic role in “depressive behaviors that persist in [the mice’s] progeny, which also show glitches in metabolism”. As one of the researches puts it, “Dad is having a much larger role in the whole process, rather than just delivering his genome and being done with it”, and a growing number of studies show that subtle change in sperm microRNAs “set the stage for a huge plethora of other effects”. In the depression study, effects persisted into the third generation of offspring. In order to rule out any form of social transmission, the researchers injected RNA collected from affected males directly into freshly fertilized eggs from untraumatized mice. “This resulted in mice with comparable depressive behaviours and metabolic symptoms — and the depressive behaviours were passed, in turn, to the next generation”. In general, however, in generations after the first offspring generation “the stressful experience did not affect the sperm microRNA”, suggesting that the originally problematic microRNA is connected with other epigenetic effects that contribute to the depressive tendencies (Hughes 2014).
-
Epigenetic modification of histones prior to zygotic genome activation
- “Marking of developmental genes by modified histones in sperm suggests a predictive role of histone marks for ZGA [zygotic genome activation]...We demonstrate here an epigenetic prepatterning of developmental gene expression”. “Early developmental instructions may thus be encoded by enrichment in specific histone marks” (Lindeman, Andersen, Reiner et al. 2011). In other words, the gametes and/or early zygote may contain histone modifications as a result of parental activity, and these modifications may help to direct the expression of developmental genes once the zygote begins its own gene transcription.
- Researchers “reduced the dimethylation of histone H3 Lys 4 (H3K4me2) in mouse sperm by overexpressing the human Lys demethylase KDM1A (also known as LSD1) specifically in the male germ line. This led to H3K4me2 loss in many developmental genes. The offspring of heterozygous transgenic males showed severe developmental defects, which were transmitted paternally through three generations, even when KDM1A was not expressed in the offspring germ line. No changes in DNA methylation were observed at CpG islands, whereas RNA profiles were altered in the sperm of transgenic males and their offspring, suggesting an important role for sperm histone methylation in transgenerational inheritance” (Baumann 2015, doi:10.1038/nrm4081).
- “Here, we show that sperm is epigenetically programmed to regulate embryonic gene expression. By comparing the development of sperm- and spermatid-derived frog embryos, we show that the programming of sperm for successful development relates to its ability to regulate transcription of a set of developmentally important genes. During spermatid maturation into sperm, these genes lose H3K4me2/3 and retain H3K27me3 marks. Experimental removal of these epigenetic marks at fertilization de-regulates gene expression in the resulting embryos in a paternal chromatin-dependent manner. This demonstrates that epigenetic instructions delivered by the sperm at fertilization are required for correct regulation of gene expression in the future embryos. The epigenetic mechanisms of developmental programming revealed here are likely to relate to the mechanisms involved in transgenerational transmission of acquired traits” (Teperek, Simeone, Gaggioli et al. 2016, doi:10.1101/gr.201541.115).
- “Recent reports probing histone modifications distribution in mouse and human sperm suggested that these epigenetic marks occurred mostly on repetitive regions of the genome rather than genes. These observations put into question the possibility that such marks would influence gene expression in embryos. By providing a functional test of the need for histone modifications for embryonic gene expression, our analysis, together with that of Siklenka et al., clearly shows that, regardless of their genomic location, sperm-delivered modified histones are important regulators of expression in future embryos” (Teperek, Simeone, Gaggioli et al. 2016, doi:10.1101/gr.201541.115).
-
X chromosome inactivation
In mammals, females possess two X chromosomes and males possess one X
chromosome and one Y chromosome. It becomes necessary for the females to
repress expression of most genes on one of their X chromosomes in order to
maintain the right balance of gene expression — that is, in order not to
express X-linked genes at twice the levels seen in males. In placental
mammals (unlike in marsupials) the choice of X chromosome to inactivate is
thought to be more or less random; about half the cells in the body repress
the maternally derived X chromosome, and half repress the paternally derived
X chromosome. About a quarter of genes on the repressed X chromosome remain
capable of expression.
Many processes involved in X chromosome inactivation “are interrelated and
function together to achieve epigenetic regulation. Recent studies show
that silencing of the inactive X chromosome involves, in addition to DNA
methylation, specific silencing histone modifications, Polycomb group
proteins, noncoding RNAs, and histone variants. All of these are likely to
be involved in transmission of the silenced state during cell division”
(Felsenfeld 2014).
(1) “The active X chromosome and inactive X chromosome (Xi) both tend to
associate with the nuclear periphery; nevertheless, different nuclear
compartments seem to influence the initiation or maintenance of XCI [X
chromosome inactivation].” (2) The silent Xi has a distinct chromatin
composition and a distinct organization of its facultative heterochromatin.
(3) The Xi possesses “a special topological structure organized into two
mega-domains or superdomains separated by the DXZ4 locus and the reduced
presence of topologically associating domains (TADs) along the chromosome
except at loci that retain transcriptional activity (escapees).” (4) “The
mechanisms behind escape of XCI remain elusive but might involve a specific
3D organization of these loci, potentially mediated by proteins such as CTCF
or YY1.” (5) “Recent studies identified a plethora of protein partners of
the Xi, including factors involved in chromosome and nuclear structure
(e.g., CIZ1, LBR, and cohesin) and phase separation mechanisms (e.g., Fus
and hnRNPA2).”
(Galupa and Heard 2018, doi:10.1146/annurev-genet-120116-024611)
A good deal is now known about the processes involved in X chromosome
inactivation (XCI); they are complex and deserve treatment here. For now,
however, you will find some mention of XCI elsewhere in this document — for
example, under Long noncoding RNAs and under
Retrotransposons.
From an article titled, “Phase Separation Drives X-Chromosome Inactivation”:
“The molecular mechanisms by which a few molecules of the long non-coding
RNA Xist silence genes on the entire X chromosome are poorly understood. New
evidence suggests that dimeric foci of Xist seed the formation of large
protein assemblies that contain a wide spectrum of proteins, such as SPEN
(SHARP), CIZ1, CELF, PTBP1 and components of Polycomb repressive complexes 1
and 2. These assemblies, each of which may contain hundreds to thousands of
molecules of proteins, extend spatially beyond each focus of Xist, which
explains how this long non-coding RNA triggers silencing across an entire
chromosome”
(Cerase, Calabrese, and Tartaglia 2022, doi:10.1038/s41594-021-00697-0).
See also Phase Transitions and Membraneless
Organelles.
“Unlike autosomal genes, X-linked genes are expressed from only one copy in
both male and female mammals. How cells increase X-linked gene expression to
match autosomal levels is unclear. New evidence suggests that lower levels
of RNA modifications on X chromosome-derived transcripts critically regulate
mRNA stability and help to balance X-to-autosome gene expression levels”
(Jachowicz 2023, doi:10.1038/s41594-023-01055-y).
Hints of the complexity of X chromosome inactivation — in particular,
“the complex combinatorial rules underlying gene silencing during X
inactivation”: “The efficiency of gene
silencing is highly variable across genes, with some genes even escaping
X chromosome inactivation (XCI)
in somatic cells. A gene's susceptibility to Xist-mediated silencing appears
to be determined by a complex interplay of epigenetic and genomic features
... The genomic distance to the Xist locus, followed by gene density and
distance to LINE elements, are the prime determinants of the speed of gene
silencing. Moreover, we find two distinct gene classes associated with
different silencing pathways: a class that requires Xist-repeat A for
silencing, which is known to activate the SPEN [transcription repressor
protein] pathway, and a second class in which genes are premarked by
Polycomb complexes and tend to rely on the B repeat in Xist for silencing,
known to recruit Polycomb complexes during XCI. Moreover, a series of
features associated with active transcriptional elongation and chromatin 3D
structure are enriched at rapidly silenced genes. Our machine-learning
approach can thus uncover the complex combinatorial rules underlying gene
silencing during X inactivation”
(Sousa, Jonkers, Syx et al. 2019, doi:10.1101/gr.245027.118).
-
Imprinting
Imprinting is an epigenetic, non-Mendelian process by which one of the two
alleles of a gene is preferentially expressed depending on whether it was
inherited from the mother or the father. Imprinted genes often occur in
clusters, and the imprinting-related locus typically has a differentially
methylated region on just one member of a chromosome pair. The imprinting
almost always involves a long noncoding RNA that, in different cases,
acts by different means. Other epigenetic processes are also known to play
roles. The many aspects of imprinting are complex, variable, and remain to
be unraveled in detail. (See also “Autosomal monoallelic expression (MAE)”
below.)
“To date, imprinted gene clusters have already provided examples of
cis-acting [roughly: local-acting] DNA sequences that are regulated
by DNA methylation, genes that are silenced by default in the mammalian
genome and require epigenetic activation to be expressed, long-range
regulatory elements that can act as insulators, and unusual long noncoding
RNAs that silence large domains of genes in cis (Barlow and
Bartolomei 2014).
The chromosomal features responsible for imprinting specific genetic
sequences are local to those sequences. However, when the imprinted
sequence is a regulatory element, that element may act at long-range on
many genes (Barlow and Bartolomei 2014).
“Although imprinted genes are repressed on one parental chromosome relative
to the other, genomic imprinting is not necessarily a silencing mechanism
and has the potential to operate at any level of gene regulation (i.e., at
the promoter, enhancers, splicing junctions, or polyadenylation sites) to
induce parental-specific differences in expression” (Barlow and Bartolomei
2014).
“The epigenetic control of imprinting mechanisms appears to be richer and
more dynamic than initially believed, with the identification of
H3K27me3-dependent imprinting control in very early development and
transient imprinting. How such transiently imprinted regions influence later
physiology requires further investigation. Among the hundreds of candidates,
expectations suggest a full range of possibilities. Some may have no
biological relevance, being silent byproducts of substantial DNA methylation
differences inherited from the parental gametes perhaps acting on adjacent
genes, some may exert an immediate effect on embryogenesis, and some will
trigger an indelible cascade with long-term phenotypic consequences”
(Tucci, Isles, Kelsey and Ferguson-Smith 2019,
doi:10.1016/j.cell.2019.01.043).
A host of regulatory possibilities:
“To control the allele-specific expression of imprinted genes in somatic
cells, gDMRs [germline differentially methylated regions] direct the
establishment of additional allele-specific epigenetic features within the
imprinted domain during development. These include secondary DMRs (also
known as somatic DMRs), which correspond mostly to gene promoters and
transcription factor binding sites, chromatin modifications, higher-order
chromatin structures (possibly resulting from CTCF–cohesin interactions) and
lncRNAs with silencing capacity for flanking imprinted genes in cis).
In other cases, imprinted gDMRs direct alternative splicing, transcription
elongation or polyadenylation site usage, which results in allele-specific
transcript isoforms. A minority of genes with parent-of-origin-dependent
expression in somatic tissues have no evident DMR in their vicinity, and
their allele-specific expression may be controlled by epigenetic features
other than DNA methylation”
(Monk, Mackay, Eggermann et al. 2019, doi:10.1038/s41576-018-0092-0).
“Many imprinted gene clusters encode microRNAs (miRNAs) and small nucleolar
RNAs (snoRNAs), which may be involved in the post-transcriptional control
of imprinted genes. These interactions might explain some of the observed
overlap in the phenotypes of different imprinting disorders”
(Monk, Mackay, Eggermann et al. 2019, doi:10.1038/s41576-018-0092-0).
“Environmental factors can influence the imprinting process. In humans,
evidence for this phenomenon derives from assisted reproductive technologies
(ARTs). Other environmental influences on imprinting centres include
nutritional status or exposure to chemical pollutants in utero”
(Monk, Mackay, Eggermann et al. 2019, doi:10.1038/s41576-018-0092-0).
“Germline-derived differential DNA methylation is the best-studied
epigenetic mark that initiates imprinting, but evidence indicates that other
mechanisms exist. Recent studies have revealed that maternal trimethylation
of H3 on lysine 27 (H3K27me3) mediates autosomal maternal allele-specific
gene silencing and has an important role in imprinted XCI [X chromosome
inactivation] through repression of maternal Xist. Furthermore, loss
of H3K27me3-mediated imprinting contributes to the developmental defects
observed in cloned embryos. This novel maternal H3K27me3-mediated
non-canonical imprinting mechanism further emphasizes the important role of
parental chromatin in development and could provide the basis for improving
the efficiency of embryo cloning”
(Chen and Zhang 2020, doi:10.1038/s41576-020-0245-9).
“DNA methylation has long been considered the primary epigenetic mediator of
genomic imprinting in mammals. Recent epigenetic profiling during early
mouse development revealed the presence of domains of trimethylation of
lysine 27 on histone H3 (H3K27me3) and chromatin compaction specifically at
the maternally derived allele, independent of DNA methylation. Within these
domains, genes are exclusively expressed from the paternally derived allele.
This novel mechanism of noncanonical imprinting plays a key role in the
development of mouse extraembryonic tissues and in the regulation of
imprinted X-chromosome inactivation, highlighting the importance of
parentally inherited epigenetic histone modifications”
(TOC blurb, https://www.cell.com/tigs/issue?pii=S0168952521X00029).
“Differences in nuclear topology precede the onset of imprinted expression
at the Peg13-Kcnk9 locus. Furthermore, the investigators provide data
in line with a model suggesting that parent-of-origin-specific topological
differences could be responsible for parent-of-origin-specific enhancer
activity and thus imprinted expression”
(Stricker 2023, doi:10.1101/gad.351216.123).
- The repressive, noncoding RNA, Kcnq1ot1 is paternally expressed in mice, and plays a repressive role in a DNA domain encompassing 14 genes on the paternal chromosome. However not all the 14 genes are repressed. Furthermore, in certain tissues at a certain stage of embryonic development, at least one of the genes previously repressed by Kcnq1ot1 begins to be expressed. This is coincident with the chromosomal locus containing the gene looping out to make contact with enhancer-like regulatory elements (Korostowski, Raval, Breuer and Engel 2011).
- A study of the mouse brain “showed parental bias in expression [of] over 1300 protein coding and putative noncoding RNAs” (Wilkinson 2010). (As of 2013 there is still some controversy about how many alleles with parentally biased expression were actually found in this work, which was conducted by Christopher Gregg et al. 2010.)
- In the same study: “Some imprinted genes show a parental bias in expression only at specific stages of development, whereas others show such expression only in certain cell types, with biallelic expression elsewhere in the brain”. Factors controlling parental influence are also sensitive to the sex of the individual and are correlated with differently spliced versions (isoforms) of the gene products from a given gene. A further complication: “In the embryonic mouse brain, there is an overall preferential maternal contribution to gene expression, which switches to a preferential paternal contribution in the adult...there is evidence for a preponderance of candidate autosomal imprinted gene loci in females that is present in the hypothalamus but not in other brain regions”. “The main message from the new findings...is that far from being some arcane sideshow, parental bias in gene expression constitutes a major component of epigenetic regulation in the mammalian brain” (Wilkinson 2010).
- “While studies have focused on determining a [DNA] sequence signature that alone could distinguish imprinted regions from the rest of the genome, recent reports do not support such a hypothesis. Rather, it is becoming clear that features such as transcription, histone modifications and higher order chromatin are employed either individually or in combination to set up parental imprints” (Abramowitz and Bartolomei 2012).
- Regarding the imprinting of noncoding RNA: an investigation of the role of imprinting in human embryogenesis “suggests that imprinting is...important in the regulation of miRNAs...Thus, alleles from a specific parent may either directly regulate cell differentiation or indirectly control the biological processes by inhibiting the expression of target genes” (Stelzer, Yanuka and Benvenisty 2011).
- In general, it needs to be recognized that imprinting can produce parent-specific effects reaching far beyond the immediately imprinted genes. If the imprinted gene is a transcription factor, then the imprinting affects all the genes influenced by that transcription factor. Likewise, miRNAs are often part of imprinted gene clusters, and “imprinted microRNAs...impose a parental specific modulation of gene expression of their target genes” (Robson, Eaton, Underhill et al. 2011). A single miRNA can affect very many targets.
- “The ubiquitin protein E3A ligase gene (UBE3A) gene is imprinted with maternal-specific expression in neurons and biallelically expressed in all other cell types. Both loss-of-function and gain-of-function mutations affecting the dosage of UBE3A are associated with several neurodevelopmental syndromes and psychological conditions, suggesting that UBE3A is dosage-sensitive in the brain ... Overall, we found no correlation between the imprinting status and dosage of UBE3A. Importantly, we found that maternal Ube3a protein levels increase in step with decreasing paternal Ube3a protein levels during neurogenesis in mouse, fully compensating for loss of expression of the paternal Ube3a allele in neurons ... we propose that imprinting of UBE3A does not function to reduce the dosage of UBE3A in neurons but rather to regulate some other, as yet unknown, aspect of gene expression or protein function” (Hillman, Christian, Doan et al. 2017, doi:10.1186/s13072-017-0134-4).
- “We recently discovered that, like DNA methylation, oocyte-inherited H3K27me3 can also serve as an imprinting mark in mouse preimplantation embryos. In this study, we found H3K27me3 is strongly biased toward the maternal allele with some associated with DNA methylation–independent paternally expressed genes in human morulae” (Zhang, Chen, Yin et al. 2019, doi:10.1101/gad.323105.118).
-
Parentally modulated DNA methylation and other epigenetic effects
There is intense investigation today of inherited epigenetic effects, where
parental lifestyle, diet, chemical exposure and other such factors result
in traits passed through one or more generations. The general idea is that
the heritable traits in question are not caused by DNA mutations, but
rather result from epigenetic modifications that influence gene expression.
While I do not discuss transgenerational epigenetic inheritance as such
here, some of the processes involved are covered elsewhere in this
document.
“Stability of the epigenetic landscape underpins maintenance of the
cell-type-specific transcriptional profile. As one of the main repressive
epigenetic systems, DNA methylation has been shown to be important for
long-term gene silencing; its loss leads to ectopic and aberrant
transcription in differentiated cells and cancer. The developing mouse germ
line endures global changes in DNA methylation in the absence of widespread
transcriptional activation ... we show that following DNA demethylation the
gonadal primordial germ cells undergo remodelling of repressive histone
modifications, resulting in a sex-specific signature in mice. We further
demonstrate that Polycomb has a central role in transcriptional control in
the newly hypomethylated germline genome as the genetic loss of Ezh2
[part of the Polycomb Repressive Complex 2, or PRC2] leads to aberrant
transcriptional activation, retrotransposon derepression and dramatic loss
of developing female germ cells. This sex-specific effect of Ezh2
deletion is explained by the distinct landscape of repressive modifications
observed in male and female germ cells”
(Huang, Wang, Vazquez-Ferrer et al. 2021, doi:10.1038/s41586-021-04208-5).
-
Promoters
[This section, along with “Pre-initiation complex” and “Transcription factors” below, constitutes a main part of “classical” gene regulation. I have omitted much basic information from these sections, and what remains is rather fragmentary, mostly pointing to other sections of this document.]
The promoter of a gene is a variously defined region most commonly found
immediately upstream of the gene’s transcription start site. However, a
promoter can also be found within a gene, or at some remove from it. In
any case, it is considered a regulatory region, and a mind-numbing array of
elements (such as DNA-binding proteins) and processes (such as histone
modifying activities) come to bear upon it in a way that modulates
expression of the associated gene or genes.
“Recent studies show that the number of bidirectional promoters (BPs) in the
human genome is much larger than previously anticipated. ... Recent studies
discuss two types of bidirectional promoters. The first type concerns
transcription of two RNAs in opposite direction from one core promoter,
i.e., one promoter leads to bidirectional transcription [5, 9, 10]. In the
second type, transcriptional initiation of both RNAs occurs at two distinct
core promoters that are close to each other, but are oriented in reverse
direction, thus sometimes termed divergent bidirectional promoters”
(Ardakani, Kattler, Nordström et al. 2018, doi:10.1186/s13072-018-0236-7).
Context matters:
“Transcription is a stochastic process involving extended periods of
inactivity interspersed with bursts of RNA synthesis. ... Now, Larsson et
al. demonstrate that enhancers predominantly modulate the frequency of
transcription bursts whereas core promoters affect their size.” “Gene
length (but not the length of the spliced mRNA) inversely correlated with
burst size but not with burst frequency. The presence of TATA elements in
gene promoters increased burst size, which was further enhanced by the
presence of initiator elements, although these had no effect on their own.
Interestingly, the effects of the different core-promoter elements on burst
size had distinct gene length dependencies; for example, gene length had a
greater effect on burst frequencies for core promoters containing TATA
elements than those lacking them.” “Importantly, cell-type-dependent
differential gene expression seemed to result mostly from differences in
burst frequency rather than burst size. Burst frequency correlated strongly
with enhancer activity” (Otto 2019, doi:10.1038/s41580-019-0100-z).
Regarding a survey of cancerous and normal tissues:
“Most human protein-coding genes are regulated by multiple, distinct
promoters, suggesting that the choice of promoter is as important as its
level of transcriptional activity ... Here, we [demonstrate] that
alternative promoters are a major contributor to context-specific regulation
of transcription. We find that promoters are deregulated across tissues,
cancer types, and patients, affecting known cancer genes and novel
candidates. For genes with independently regulated promoters, we demonstrate
that promoter activity provides a more accurate predictor of patient
survival than gene expression. Our study suggests that a dynamic landscape
of active promoters shapes the cancer transcriptome, opening new diagnostic
avenues and opportunities to further explore the interplay of regulatory
mechanisms with transcriptional aberrations in cancer”
(Demircioğlu, Cukuroglu Kindermans et al. 2019,
doi:10.1016/j.cell.2019.08.018).
“Precise spatiotemporal control of gene expression during normal development
and cell differentiation is achieved by the combined action of proximal
(promoters) and distal (enhancers) cis-regulatory elements. Recent studies
have reported that a subset of promoters, termed Epromoters, works also as
enhancers to regulate distal genes. This new paradigm opened novel questions
regarding the complexity of our genome and raises the possibility that
genetic variation within Epromoters has pleiotropic effects on various
physiological and pathological traits by differentially impacting multiple
proximal and distal genes. Here, we discuss the different observations
pointing to an important role of Epromoters in the regulatory landscape and
summarize the evidence supporting a pleiotropic impact of these elements in
disease. We further hypothesize that Epromoter might represent a major
contributor to phenotypic variation and disease”
(Malfait, Wan and Spicuglia 2023, doi:10.1002/bies.202300012).
- “The immediate role of the promoter is to bind and correctly position the transcription initiation complex. ... In eukaryotes, RNA polymerase II (RNAPII)-transcribed genes are highly heterogeneous with respect to expression level and context specificity. Therefore, their transcriptional control needs to be highly specialized and dynamic; an important part of this diversity is mediated by different classes of RNAPII promoters, which differ dramatically in their architecture, which in turn determines the promoter function and regulation type” (Lenhard, Sandelin and Carninci 2012).
- Actually, the situation does not seem all that determinate. Referring to the “nuanced” functional character of promoters, Roy and Singer (2015a, doi:10.1016/j.tibs.2015.01.007) offer this example: “The MHC class I promoter contains both a TATAA-like element and a canonical Inr. Also within the 60 base-pair promoter region is a CCAAT box and an Sp1 binding site ... Surprisingly, none of these promoter elements was essential for promoter activity or transcription initiation in vivo. All of the mutants supported transcription in vivo as well as or better than the wild type promoter. Although none was necessary for transcription, each element had a defined role. Thus, CAAT box mutations modulated constitutive expression in non-lymphoid tissues, whereas TATAA-like element mutations dysregulated transcription in lymphoid tissues. Conversely, Inr and Sp1 binding element mutations aberrantly elevated expression in both lymphoid and non-lymphoid tissues”.
- “A recent study also raises questions about the concept of a core promoter. It reports that thousands of promoters in vertebrates, including those of ubiquitously expressed genes, contain at least two TSS [transcription start site] ‘selection codes’. The first is mostly utilized in oocytes and the other in developing embryos. While the first code is dependent on a weak TATA-like sequence, the second code is dependent on the position of the H3K4me3-marked first nucleosome downstream of the TSS. These observations suggest that, rather than one open promoter architecture, multiple overlapping promoter codes dictate the expression of a ubiquitously expressed gene”. Moreover, “most mammalian genes lack canonical core promoter elements but nevertheless recruit the transcriptional machinery” (Roy and Singer 2015a, doi:10.1016/j.tibs.2015.01.007)
- “Another surprising finding from genome-wide studies is that most mammalian promoters direct transcription initiation in both directions with opposite orientation, a phenomenon termed ‘divergent’ transcription” (Roy and Singer 2015a, doi:10.1016/j.tibs.2015.01.007)
- “DNA methylation at the promoter of a gene is presumed to render it silent, yet a sizable fraction of genes with methylated proximal promoters exhibit elevated expression. [We show that] in many such cases, transcription is initiated by a distal upstream CpG island (CGI) located several kilobases away that functions as an alternative promoter. Specifically, such genes are expressed precisely when the neighboring CGI is unmethylated but remain silenced otherwise ... Overall, our study describes a hitherto unreported conserved mechanism of transcription of genes with methylated proximal promoters in a tissue-specific fashion” (Sarda, Das, Vinson and Hannenhalli 2017, doi:10.1101/gr.212050.116).
- The functional distinction between promoters and enhancers has become increasingly blurred with time, with complex, context-specific interaction between the two commonly being decisive for the particulars of gene expression. See Enhancers and silencers below.
- “Gene expression in higher eukaryotes is precisely regulated in time and space through the interplay between promoters and gene-distal regulatory regions, known as enhancers. The original definition of enhancers implies the ability to activate gene expression remotely, while promoters entail the capability to locally induce gene expression. Despite the conventional distinction between them, promoters and enhancers share many genomic and epigenomic features. One intriguing finding in the gene regulation field comes from the observation that many core promoter regions display enhancer activity. Recent [researches] have indicated that this phenomenon is common and might have a strong impact on our global understanding of genome organisation and gene expression regulation” (Medina-Rivera, Santiago-Algarra, Puthier and Spicuglia 2018, doi:10.1016/j.tibs.2018.03.004).
- “Most mammalian genes lack a well-defined core promoter element but instead contain promoter regions with characteristic epigenetic marks (both histone chromatin and DNA marks) ... Given that RNA Pol II appears to be recruited to large stretches of the genome without identifiable core promoter elements, are core-promoter elements necessary for the recruitment of the transcriptional machinery? ... Perhaps canonical core promoter elements fine-tune physiological responses for a select few genes in a developmental fashion and work in concert with epigenetic marks and other regulatory elements such as enhancers ... Moreover, given the heterogeneity of core promoter architecture, it is also likely that the eukaryotic genome utilizes many of these elements in a mix-and-match fashion, thereby increasing the regulatory capacity of transcription initiation. Given that most promoters lack canonical core promoter elements, the importance of the noncanonical promoter elements has recently been brought into light ... it is clear that the mammalian genome has taken advantage of multiple regulatory strategies to initiate and regulate the transcription of both protein-coding and protein-noncoding units. (Roy and Singer 2015a, doi:10.1016/j.tibs.2015.01.007)
- Promoters in general have been thought to offer mostly standard sequences for transcription factors, and therefore not to be heavily involved in differential gene regulation. However, two researchers working with five Drosophila species, investigated gene expression patterns during different stages of early development. They found 3973 promoters, mostly uannotated and widely associated with noncoding DNA, that drove expression during embryonic development (Batut and Gingeras 2017, doi:10.7554/eLife.29005).
- “Here we measure mRNA levels for 10,000 open reading frames (ORFs) transcribed from either an inducible or constitutive promoter. We find that the strength of cotranslational regulation on mRNA levels is determined by promoter architecture ... we identify the RNA helicase Dbp2 as the mechanism by which cotranslational regulation is reduced specifically for inducible promoters. Finally, we find that for constitutive genes, but not inducible genes, most of the information encoding regulation of mRNA levels in response to changes in growth rate is encoded in the ORF and not in the promoter. Thus, the ORF sequence is a major regulator of gene expression, and a nonlinear interaction between promoters and ORFs determines mRNA levels” (Espinar, Tamarit, Domingo and Carey 2018, doi:10.1101/gr.230458.11).
- “Many human genes have tandem promoters driving overlapping transcription, but the value of this distributed promoter configuration is generally unclear. Here we show that MICA, a gene encoding a ligand for the activating immune receptor NKG2D, contains a conserved upstream promoter that expresses a noncoding transcript. Transcription from the upstream promoter represses the downstream standard promoter activity in cis through transcriptional interference. The effect of transcriptional interference depends on the strength of transcription from the upstream promoter and can be described quantitatively by a simple reciprocal repressor function. Transcriptional interference coincides with recruitment at the standard downstream promoter of the FACT histone chaperone complex, which is involved in nucleosomal remodelling during transcription. The mechanism is invoked in the regulation of MICA expression by the physiological inputs interferon‐γ and interleukin‐4 that act on the upstream promoter. Genome‐wide analysis indicates that transcriptional interference between tandem intragenic promoters may constitute a general mechanism with widespread importance in human transcriptional regulation” (Hiron and O’Callaghan 2018, doi:10.15252/embj.201797138).
-
A few generalities
- DNA sequence of the promoter region. Sequences, with almost unlimited variation, have been more or less hazily classified into various types, corresponding to different classes of genes and gene expression patterns (Lenhard, Sandelin and Carninci 2012). There are many sequence elements, such as TATA boxes, CpG islands, downstream core elements, DNA recognition elements, and so on, that appear in different sorts of promoters in different combinations.
- DNA and histone tail modifications. “Epigenetic signals — namely, histone and DNA modifications — have been associated with promoter class and functional state” (Lenhard, Sandelin and Carninci 2012). For information on “DNA methylation” and “Histone tail modifications” in general, see those topics below.
- Nucleosomes — presence and positioning at promoters “Different classes of promoters seem to have different patterns of nucleosome occupancy and precision of positioning” (Lenhard, Sandelin and Carninci 2012). And apart from a consideration of nucleosomes in relation to classes of promoters, nucleosome-related modulation of gene expression at individual genes is hugely complex. See the treatment of nucleosome positioning and nucleosome remodeling below.
- “We profiled and compared transcriptional and regulatory element activities across five tissues of Caenorhabditis elegans, covering ∼90% of cells. We find that the majority of promoters and enhancers have tissue-specific accessibility, and we discover regulatory grammars associated with ubiquitous, germline, and somatic tissue–specific gene expression patterns. In addition, we find that germline-active and soma-specific promoters have distinct features. Germline-active promoters have well-positioned +1 and −1 nucleosomes associated with a periodic 10-bp WW signal (W = A/T). Somatic tissue–specific promoters lack positioned nucleosomes and this signal, have wide nucleosome-depleted regions, and are more enriched for core promoter elements, which largely differ between tissues” (Serizay, Dong, Jänes et al. 2020, doi:10.1101/gr.265934.120).
- Chromatin remodeling. If nucleosomes play a regulatory role at promoters, the usual infinite explanatory regress leads next to the question, “What regulates the nucleosomes?” In addition to histone tail modifications listed above (and discussed in wider contexts below), there are chromatin remodeling protein complexes that can shift the position of nucleosomes, thereby tending to enhance or repress gene expression from the given promoter.
- Long-range interactions. Again, long-range interactions between promoters and regulatory elements such as enhancers vary between different classes of genes, and this variation is linked to other factors such as nucleosome positioning, the histone modification or DNA methylation of the enhancers, and the factors affecting chromosome looping and chromosome location within the nucleus.
- Retrotransposons as promoters. Remarkably, it’s been found that retrotransposons, widely dismissed as “parasitic DNA” (see REPETITIVE AND TRANSPOSABLE DNA below), can be recruited as promoters for the expression of tissue-specific, noncoding RNAs. Studies “have identified more than 200,000 human retrotransposon-driven TSSs [transcription start sites], which are expressed at low to moderate levels. ... Frequently, retrotransposon-mediated TSSs start upstream of typical mRNA promoter regions, and they produce RNAs that are transcribed towards the downstream genes”. Evidence suggests that “these retrotransposon promoters have complex transcriptional regulation. ... RNAs that are derived from these promoters often lack the polyadenylation tail and are often localized in the nucleus, suggesting that they may have a role in transcriptional regulation and/or nuclear organization” (Lenhard, Sandelin and Carninci 2012). “We conclude that retrotransposon transcription has a key influence upon the transcriptional output of the mammalian genome” (Faulkner, Kimura, Daub et al. 2009).
-
Promoter activation kinetics
- Promoters may be slower or faster to respond to binding by a given transcription factor, and many transcription factors (and signaling processes in general) exhibit time-dependent behavior. “In response to oscillatory transcription factor inputs, slow promoters that are activated by an input pulse cannot fully return back to the inactive state when the next input pulse occurs, so they begin to increase expression from a higher starting point. This ‘head-start’ effect, which is more marked in response to high frequency input, results in a nonlinear relationship between response level and input frequency. By contrast, genes with fast promoter kinetics generate isolated expression responses to each transcription factor pulse; therefore the response is proportional to the input frequency” (Hao and O’Shea 2012; see also Hansen and O’Shea 2013). And so different transcription factor dynamics can elicit different expression patterns from different genes. (See “Additional dynamic aspects of transcription factor activity” below.) And, of course, such things as nucleosome positioning and stability will likely play a role in influencing promoter kinetics: “Examination of nucleosome structure on three of the seven promoters (one slow and two fast) showed that, indeed, chromatin remodeling occurred more quickly at fast promoters” (Moody and Batchelor 2013, reporting on Hansen and O’Shea 2013).
- “Mammalian gene expression is inherently stochastic, and results in discrete bursts of RNA molecules that are synthesized from each allele ... We show that core promoter elements affect burst size and uncover synergistic effects between TATA and initiator elements, which were masked at mean expression levels. Notably, we provide transcriptome-wide evidence that enhancers control burst frequencies, and demonstrate that cell-type-specific gene expression is primarily shaped by changes in burst frequencies. Together, our data show that burst frequency is primarily encoded in enhancers and burst size in core promoters” (Larsson, Johnsson, Hagemann-Jensen et al. 2018, doi:10.1038/s41586-018-0836-1).
-
Dynamics of RNA polymerase II
- The behavior of RNA polymerase at the promoter — for example, it’s being paused there as opposed to quickly moving into elongation phase — has effects upon gene expression. See “RNA polymerase pausing and release” under DECISION-MAKING DURING TRANSCRIPTION below.
- Kinetic promoter proofreading “[RNA] Polymerase structure is permissive for abortive initiation, thereby setting a lower limit on polymerase-promoter complex lifetime and allowing the dissociation of nonspecific [initiation] complexes. Abortive initiation may be viewed as promoter proofreading, and the structural transitions as checkpoints for promoter control” (Liu, Bushnell, Silva et al. 2011).
-
Complexities
- “Gene transcription is highly regulated through the surrounding regulatory environment, such as the formation of enhancer-promoter/promoter-promoter contacts, dynamic molecular clustering of transcription machineries, and possibly through RNA-protein interactions within the nucleus. In recent years, many intriguing hypotheses have been proposed to explain the molecular mechanisms of enhancer function, including the liquid-liquid phase separation model. Yet, it is still challenging to consolidate all the possible scenarios into a single clear picture. In this review article, we discussed long-range cross-regulation at the scyl/chrb and GluRIA/GluRIB loci as a representative example, but the mode of cross-regulation of functionally related distant genes through the formation of promoter-promoter loops appears to be different from locus to locus. Therefore, our current understanding is far from establishing a general model that explains the functionalities of this newly recognized mechanism. Another key question that remains to be addressed is how the specificity of enhancer-promoter/promoter-promoter contacts is determined in the context of animal development. Recent studies suggest that tethering elements and promoter CpG methylation act as key layers in defining the regulatory connectivity within the complex genome. However, it remains mostly unclear how specific pairs of tethering elements find each other over large genomic distances to establish new regulatory loops” (Makino and Fukaya 2024, doi:10.1002/bies.202400101).
- In sum: “Only a minority of promoters fit the ‘classical’ model of transcriptional initiation and regulation: tissue-specific ... promoters, which have most of their regulatory elements close to the transcription start site and are controlled locally. A much larger fraction of genes is regulated by broad promoters with activity that seems to be more influenced by the epigenomic context and less so by sequence-specific transcription factors” (Lenhard, Sandelin and Carninci 2012).
-
Pre-initiation complex
The pre-initiation complex (PIC) consists of a group of multi-subunit
protein complexes known as general (or basal or core)
transcription factors, including RNA polymerase, that come together
on a gene promoter as a preparatory step for gene transcription. This
complex was formerly thought to consist of standard parts assembled in a
fixed, step-by-step manner, leading to gene expression. But now it is
being recognized that there are almost endless possibilities for variable
combination of PIC subunits and variable interaction with gene promoters,
constituting the PIC as a highly flexible and context-sensitive set of
factors in gene regulation (Goodrich and Tjian 2010).
- “The first step in PIC assembly is binding of TFIID, a multisubunit complex consisting of TATA-box-binding protein (TBP) and a set of 14 TBP-associated factors (TAFs). Transcription then proceeds through a series of steps, including promoter melting, clearance, and escape, before fully functional PolII elongation is achieved. Alternative core promoter complexes may help to maintain specific transcriptional programmes in terminally differentiated cell types”. (Vernimmen and Bickmore 2015, doi:10.1016/j.tig.2015.10.004)
- “Models of transcription regulation view this as a cycle, in which complete PIC assembly is stimulated only once. After PolII escapes from the promoter, TFIID, TFIIE, TFIIH and the mediator complex remain on the core promoter; subsequent reinitiation then only requires de novo recruitment of subcomplexes comprising PolII–TFIIF and TFIIB. The various steps of PIC assembly on a core promoter can occur with different timings during differentiation”. (Vernimmen and Bickmore 2015, doi:10.1016/j.tig.2015.10.004)
-
“However, even with cooperative interactions between components, the
assembly of >70 protein subunits for a eukaryotic PIC seems like an
impossible task. Thus, it is no surprise that only one in 90 collisions
of Pol II with the DNA template is thought to result in productive
elongation (Darzacq et al. 2007) ... The difficulty in fully activating a
gene could help to prevent spurious undesired transcription and has also
been suggested as a possible cause of transcriptional bursting” (Chen and
Larson 2016, doi:10.1101/gad.281725.116).
(This concern about the “difficulty” of such-and-such a molecular achievement is encountered all too often in the literature, and is strangely anthropomorphic. Since cells seem to get the job done just about right, where is the difficulty? What drives this kind of talk is apparently a naive picture of what would be the efficient way to do things, and this in turn is owing to a simplistic, machine-like view of the tasks at hand. But if you review anything like the complete contents of the notes you are now reading, it is obvious that we have hardly begun to grasp how any particular activity is interwoven with numerous others. We still have little clue about the various processes to which RNA Pol II is contributing when it “bounces off” the DNA template.)
- “Eukaryotic protein‐coding genes are typically classified into two groups: those with expression regulated by specific signals versus the relatively constant “housekeeping” genes. Although these differences are associated with alternative modes of RNA polymerase II (RNAP II) pre‐initiation complex (PIC) assembly, a role for gene‐specific activators in controlling “regulatability” has been difficult to rule out. To address this question, de Jonge et al (2017) studied a group of genes controlled by a common activator but dependent on [PIC factors] TFIID or SAGA and found that the magnitude of regulation strongly correlates with the mechanism of PIC assembly” (Kubik, Bruzzone and Shore 2017, doi:10.15252/embj.201696152).
- “Nuclear small RNA pathways safeguard genome integrity by establishing transcription-repressing heterochromatin at transposable elements. This inevitably also targets the transposon-rich source loci of the small RNAs themselves. How small RNA source loci are efficiently transcribed while transposon promoters are potently silenced is not understood. Here we show that, in Drosophila, transcription of PIWI-interacting RNA (piRNA) clusters—small RNA source loci in animal gonads—is enforced through RNA polymerase II pre-initiation complex formation within repressive heterochromatin. This is accomplished through Moonshiner, a paralogue of a basal transcription factor IIA (TFIIA) subunit, which is recruited to piRNA clusters via the heterochromatin protein-1 variant Rhino. Moonshiner triggers transcription initiation within piRNA clusters by recruiting the TATA-box binding protein (TBP)-related factor TRF2, an animal TFIID core variant. Thus, transcription of heterochromatic small RNA source loci relies on direct recruitment of the core transcriptional machinery to DNA via histone marks rather than sequence motifs, a concept that we argue is a recurring theme in evolution” (Andersen, Tirian, Vunjak and Brennecke 2017, doi:10.1038/nature23482).
- Transcription complexes and disordered protein domains: “Many components of eukaryotic transcription machinery—such as transcription factors and cofactors including BRD4, subunits of the Mediator complex, and RNA polymerase II — contain intrinsically disordered low-complexity domains. Now a conceptual framework connecting the nature and behavior of their interactions to their functions in transcription regulation is emerging. Chong et al. found that low-complexity domains of transcription factors form concentrated hubs via functionally relevant dynamic, multivalent, and sequence-specific protein-protein interaction. These hubs have the potential to phase-separate at higher concentrations. Indeed, Sabari et al. showed that at super-enhancers, BRD4 and Mediator form liquid-like condensates that compartmentalize and concentrate the transcription apparatus to maintain expression of key cell-identity genes. Cho et al. further revealed the differential sensitivity of Mediator and RNA polymerase II condensates to selective transcription inhibitors and how their dynamic interactions might initiate transcription elongation” (Chong, Dugast-Darzacq, Liu et al, 2018, doi:10.1126/science.aar2555).
-
Tata-binding protein (TBP)
The Tata-binding protein (part of the TFIID complex; see below) nucleates the pre-initiation complex on the promoter of many genes. It severely bends the DNA, loosening the two strands of the double helix and preparing the way for the binding of the remaining memebers of the PIC.- Researchers had assumed that TBP was stably bound to the gene promoter during successive transcription runs. However, it now appears that “a highly mobile TBP population is critical for transcriptional regulation on a global scale”. “The entire (or nearly entire) TBP pool is rapidly recycled, leading to rapid redistribution of TBP among chromatin binding sites”. When the cycles of assembly and diassembly of the TBP-containing complexes on promoters are disrupted, the transcription process fails to run to completion, resulting in aberrant RNA transcripts. (Poorey, Sprouse, Wells et al. 2010).
-
General transcription factors: TFIIA, TFIIB, TFIID, TFIIE, TFIIF, TFIIH
[To Do: try to reduce the huge variety of regulatory processes here to a brief, presentable summary. This is a vastly complex area and should constitute a major section of this document.] -
Mediator complex
- The Mediator complex forms part of the pre-initiation complex. “The mechanisms by which Mediator regulates gene expression remain poorly understood, in part because the structure of Mediator and even its composition can change, depending upon the promoter context. Combined with the sheer size of the human Mediator complex (26 subunits, 1.2 MDa), this structural adaptability bestows seemingly unlimited regulatory potential within the complex...it is also evident that Mediator performs both general and gene-specific roles to regulate gene expression” (Taatjes 2010). It plays these roles “at each stage of transcription, from the recruitment of pol II [RNA polymerase II] to genes in response to many signals, to controlling pol II activity during transcription initiation and elongation” (Conaway and Conaway 2011).
- The Mediator is a “key regulator of protein-coding genes...multiple pathways that are responsible for homeostasis, cell growth and differentiation converge on the Mediator through transcriptional activators and repressors that target one or more of the almost 30 subunits of this complex. Besides interacting directly with RNA polymerase II, Mediator has multiple functions and can interact with and coordinate the action of numerous other co-activators and co-repressors, including those acting at the level of chromatin. These interactions ultimately allow the Mediator to deliver outputs that range from maximal activation of genes to modulation of basal transcription to long-term epigenetic silencing” (Malik and Roeder 2010).
- Mediator can also have tissue-specific effects: “Adding yet another degree of complexity, members of the same transcription factor family can target different Mediator subunits to activate transcription of the same gene, through the same promoter elements, in different cell types. (Conaway and Conaway 2011).
- “The large multiprotein Mediator complex can act as a bridge between transcription activators and components of the PIC. It appears to play important roles in many steps of transcription, including PIC formation and the transition to elongation. Mediator is >1 MDa in size and >30 nm in length, with distinct structural modules and a flexible structure that changes in response to the binding of different TFs. TF binding seems to induce a conformational change in Mediator that facilitates PolII binding. Different TFs bind different Mediator subunits, and Mediator complexes that lack a specific subunit can still activate transcription in response to TFs that bind to other subunits. Therefore, among other proteins (e.g., CTCF and cohesin complex) ... Mediator provides an important bridge for integrating information coming from different signalling pathways. Mediator might also provide an important binding surface for noncoding RNAs, including enhancer RNAs”. (Vernimmen and Bickmore 2015, doi:10.1016/j.tig.2015.10.004)
- “We revealed an essential function of the Mediator middle module exerted through its Med10 subunit, implicating a key interaction between Mediator and TFIIB. We showed that this Mediator–TFIIB link has a global role on PIC assembly genome-wide. Moreover, the amplitude of Mediator's effect on PIC formation is gene-dependent and is related to the promoter architecture in terms of TATA elements, nucleosome occupancy, and dynamics” (Eychenne, Novikova, Barrault et al. 2016, doi:10.1101/gad.285775.116).
- “Instead of eliminating activity as expected, the depletion of individual [Mediator] subunits caused a modest decrease in transcription. Only when all three (head, middle, and tail) modules of Mediator were simultaneously inactivated was transcription abrogated. Furthermore, different Mediator modules promoted RNA polymerase II in different ways, and Mediator was not found in the preinitiation complex. This result questions the classic model of Mediator bridging enhancers and promoters and begs for answers about how Mediator activates transcription” (Mao 2017, https://science.sciencemag.org/content/357/6350/twil).
-
“Mediator structure and function is completely altered upon binding
the Mediator kinase module, a multi-subunit complex that contains CDK8
or its vertebrate-specific paralog CDK19. Here, we review the
mechanisms by which the Mediator kinase module controls pol II
transcription, emphasizing its impact on TF [transcription factor]
activity, pol II elongation, enhancer function, and chromatin
architecture. We also highlight how the Mediator kinase module
integrates signaling pathways with transcription to enable rapid,
stimulus-specific responses, as well as its links to human disease.”
“Through kinase-dependent mechanisms, CDK8 and/or CDK19 regulate TF function to help ‘reprogram’ gene expression patterns in response to a stimulus or developmental cues. The Mediator kinase module also functions in kinase-independent ways, through Mediator binding, which blocks Mediator-pol II interaction yet appears to promote post-initiation events, such as pol II pause release or elongation. The complexity of the pol II transcription machinery and cell signaling networks presents many opportunities for new discoveries, but also many challenges. Cell type and cell context (e.g., oxidative stress or growth factor induction) will remain important considerations in future work, as the set of active TFs will change in each case” (Luyties and Taatjes 2022, doi:10.1016/j.tibs.2022.01.002).
- “Mediator is a coregulatory complex that plays essential roles in multiple processes of transcription regulation. One of the human Mediator subunits, MED26, has a role in recruitment of the super elongation complex (SEC) to polyadenylated genes and little elongation complex (LEC) to non-polyadenylated genes, including small nuclear RNAs (snRNAs) and replication-dependent histone (RDH) genes. MED26-containing Mediator plays a role in 3′ Pol II pausing at the proximal region of transcript end sites in RDH genes through recruitment of Cajal bodies (CBs) to histone locus bodies (HLBs). This finding suggests that Mediator is involved in the association of CBs with HLBs to facilitate 3′ Pol II pausing and subsequent 3′-end processing by supplying 3′-end processing factors from CBs. Thus, we argue the possibility that Mediator is involved in the organization of nuclear bodies to orchestrate multiple processes of gene transcription” (table of contents blurb for Suzuki, Furugori, Ryota Abe et al. 2023, doi:10.1002/bies.202200178).
-
Transcription factors (other than general transcription factors)
[General transcription factors are discussed — or,
rather, just mentioned — above.]
“Most complex trait-associated variants are located in non-coding regulatory
regions of the genome, where they have been shown to disrupt transcription
factor (TF)-DNA binding motifs. Variable TF-DNA interactions are therefore
increasingly considered as key drivers of phenotypic variation. However,
recent genome-wide studies revealed that the majority of variable TF-DNA
binding events are not driven by sequence alterations in the motif of the
studied TF. This observation implies that the molecular mechanisms
underlying TF-DNA binding variation and, by extrapolation, inter-individual
phenotypic variation are more complex than originally anticipated. Here, we
summarize the findings that led to this important paradigm shift and review
proposed mechanisms for local, proximal, or distal genetic variation-driven
variable TF-DNA binding”
(Deplancke, Alpern and Gardeux 2016, doi:10.1016/j.cell.2016.07.012).
“New genomic analyses indicate that pioneer transcription factors can sample
a diverse repertoire of common binding sites among different cell types and
become enriched where they cooperate with other factors specific to each
cell. Pioneer-factor binding is mechanistically separate from, and is
necessary for, subsequent phenomena of chromatin opening and epigenetic
memory in vivo” (Zaret 2018, doi:10.1038/s41588-017-0038-z).
“Transcriptional silencing may not necessarily depend on the continuous
residence of a sequence‐specific repressor at a control element and may act
via a “hit and run” mechanism ... To explore this possibility, erythroid
gene promoters that are regulated directly by GATA1 in an inducible system
are analyzed. It is found that many regulated genes are bound immediately
after induction of GATA1 but the residency of GATA1 decreases over time,
particularly at repressed genes. Furthermore, it is shown that the
repressive mark H3K27me3 is seldom associated with bound repressors,
whereas, in contrast, the active (H3K4me3) histone mark is overwhelmingly
associated with TF [transcription factor] binding. It is hypothesized that
during cellular differentiation and development, certain genes are silenced
by repressive TFs that subsequently vacate the region. Catching such
repressor TFs in the act of silencing via assays such as ChIP‐seq is thus a
temporally challenging prospect”
(Shah, Funnell, Quinlan and Crossley 2019, doi:10.1002/bies.201900041).
Transcription factor interaction and post-translational modification.
“Embryonic stem (ES) cells are regulated by a network of transcription
factors that maintain the pluripotent state. Differentiation relies on
down-regulation of pluripotency transcription factors disrupting this
network. While investigating transcriptional regulation of the pluripotency
transcription factor Kruppel-like factor 4 (Klf4), we observed that
homozygous deletion of distal enhancers caused a 17-fold decrease in Klf4
transcript but surprisingly decreased protein levels by less than twofold,
indicating that posttranscriptional control of KLF4 protein overrides
transcriptional control. The lack of sensitivity of KLF4 to transcription is
due to high protein stability (half-life >24 h). This stability is
context-dependent and is disrupted during differentiation, as evidenced by a
shift to a half-life of <2 h. KLF4 protein stability is maintained
through interaction with other pluripotency transcription factors (NANOG,
SOX2, and STAT3) that together facilitate association of KLF4 with RNA
polymerase II. In addition, the KLF4 DNA-binding and transactivation
domains are required for optimal KLF4 protein stability. Posttranslational
modification of KLF4 destabilizes the protein as cells exit the pluripotent
state ... These data indicate that the core pluripotency transcription
factors are integrated by posttranslational mechanisms to maintain the
pluripotent state ...”
(Dhaliwal, Abatti and Mitchell 2019, doi:10.1101/gad.324319.119).
“Transcription factors (TFs) bind to specific DNA motifs to regulate the
expression of target genes. To reach their binding sites, TFs diffuse in 3D
and perform local motions such as 1D sliding, hopping, or intersegmental
transfer. TF–DNA interactions depend on multiple parameters, such as the
chromatin environment, TF partitioning into distinct subcellular regions,
and cooperativity with other DNA-binding proteins”
(Suter 2020, doi:10.1016/j.tcb.2020.03.003).
“Pioneer factors are transcriptional regulators with the capacity to bind
inactive regions of chromatin and induce changes in accessibility that
underpin cell fate decisions. The FOXA family of transcription factors is
well understood to have pioneer capacity. Indeed, researchers have uncovered
numerous examples of FOXA-dependent epigenomic modulation in developmental
and disease processes. Despite the presence of FOXA being essential for
correct epigenetic patterning, the need for continued FOXA presence
postchromatin modulation has been debated. [A recent study shows] that the
tissue-specific ablation of FOXA1/2/3 in the adult mouse liver results in
the collapse of the epigenetic profile that maintains the hepatic gene
expression profile. Thus, FOXA functions as a key, opening regions of
chromatin during development, and as a doorstep, maintaining the established
euchromatic structure in adult tissue”
(Heslop and Duncan 2020, doi:10.1101/gad.340570.120).
“Advances in fluorescence microscopy have made it possible to visualize
real-time TF dynamics in living cells, leading to two intriguing
observations: first, most TFs contact chromatin only transiently; and
second, TFs can assemble into clusters through their intrinsically
disordered regions. These findings suggest that highly dynamic events and
spatially structured nuclear microenvironments might play key roles in
transcription regulation that are not yet fully understood. The emerging
model is that while some promoters directly convert TF-binding events into
on/off cycles of transcription, many others apply complex regulatory layers
that ultimately lead to diverse phenotypic outputs”
(Lu and Lionnet 2021, doi:10.1101/cshperspect.a040949).
[Regarding an investigation into the complexity of neuron specification
regulatory networks in Caenorhabditis elegans relative to nine
different neuron types:] “We identified 91 TF candidates to be required for
correct generation of these neuron types, of which 28 were confirmed by
mutant analysis. We found that correct reporter expression in each
individual neuron type requires at least nine different TFs. Individual
neuron types do not usually share TFs involved in their specification but
share a common pattern of TFs belonging to the five most common TF families:
homeodomain (HD), basic helix loop helix (bHLH), zinc finger (ZF), basic
leucine zipper domain (bZIP), and nuclear hormone receptors (NHR). HD TF
members are overrepresented, supporting a key role for this family in the
establishment of neuronal identities. These five TF families are also
prevalent when considering mutant alleles with previously reported neuronal
phenotypes in C. elegans, Drosophila, and mouse. In addition
... combined TF binding sites for these five TFs constitute a
cis-regulatory signature enriched in the regulatory regions of
dopaminergic effector genes”
(Jimeno-Martín, Sousa, Brocal-Ruiz et al. 2022, doi:10.1101/gr.275623.121).
Overall, the dominating feature of TF dynamics is that they follow a
distributed interaction principle, apparent at many scales. First, in
stoichiometric fuzzy complexes, multiple weak interaction sites between two
partners rapidly exchange without complex dissociation. Second, multivalent
interactions distributed across IDRs [intrinsically disordered regions]
ensure the formation of nonstoichiometric clusters. Finally, enhancers favor
multiple weak TF-binding motifs over high affinity ones to ensure expression
specificity. This unifying principle offers many advantages needed for
regulatory function, particularly robustness, tunability, and
responsiveness”
(Lu and Lionnet 2021, doi:10.1101/cshperspect.a040949).
“Pioneer factors are transcription factors with the unique ability to
initiate opening of closed chromatin. The stability of cell identity relies
on robust mechanisms that maintain the epigenome and chromatin accessibility
to transcription factors. Pioneer factors counter these mechanisms to
implement new cell fates through binding of DNA target sites in closed
chromatin and introduction of active-chromatin histone modifications,
primarily at enhancers. As master regulators of enhancer activation,
pioneers are thus crucial for the implementation of correct cell fate
decisions in development”
(Balsalobre and Drouin 2022, doi:10.1038/s41580-022-00464-z).
“Transcription factors (TFs) are considered to have two functional domains —
a DNA-binding domain, with which they bind to enhancers and promoters, and a
protein-binding domain. Although non-coding RNA molecules are produced at
enhancers and promoters in proximity to TF binding sites, TFs do not have
‘canonical’ RNA-binding domains, and the role of these RNAs in transcription
is debated. Oksuz, Henninger et al. now show that various TFs indeed bind
RNA — through a specific motif — and that this interaction can promote
transcription and has a role in development and disease”
(Zlotorynski 2023, doi:10.1038/s41580-023-00643-6).
- Transcription factors — proteins that bind to specific DNA sequences — are the classic regulators of gene expression. Most transcription factors (we now know) bind to DNA at thousands of loci. They often play a direct role in recruiting other proteins essential for transcription or repression of a gene — co-activators, co-repressors, basal (general) transcription factors, and so on.
- Transcription factors, like all protein regulators of transcription, are themselves regulated via endlessly complex pathways. And their binding to a specific DNA locus does not at all mean there will be an associated and detectable change in a gene’s transcription. It is, in fact, often hard to know which gene might be expected to respond the the factor. “Transcription factors generally act in a context-dependent, combinatorial manner” (Dowell 2010).
- Role of dyamic changes in transcription factor form (conformation): “The glucocorticoid receptor (GR) is a constitutively expressed transcriptional regulatory factor that controls many distinct gene networks, each uniquely determined by particular cellular and physiological contexts. The precision of GR-mediated responses seems to depend on combinatorial, context-specific assembly of GR-nucleated transcription regulatory complexes at genomic response elements. In turn, evidence suggests that context-driven plasticity is conferred by the integration of multiple signals [such as DNA sequences, ligands, post-translational modifications and other transcription regulators], each serving as an allosteric effector of GR conformation, a key determinant of regulatory complex composition and activity. This structural and mechanistic perspective on GR regulatory specificity is likely to extend to other eukaryotic transcriptional regulatory factors” (Weikum, Knuesel, Ortlund and Yamamoto 2017, doi:10.1038/nrm.2016.152).
-
A few examples of the regulation of transcription factors:
- Post-translational modifications play a role in the functioning of transcription factors. For example, “The functions of the FoxO family proteins, in particular their transcriptional activities, are modulated by post-translational modifications (PTMs), including phosphorylation, acetylation, ubiquitination, methylation and glycosylation. These PTMs occur in response to different cellular stresses, which in turn regulate the subcellular localization of FoxO family proteins, as well as their half-life, DNA binding, transcriptional activity and ability to interact with other cellular proteins” (Zhao, Wang and Zhu 2011).
- Sumoylation (attachment of a small protein called SUMO) is another post-translational modification. Many repressive factors such as histone deacetylases and polycomb-related repressors “are more effectively recruited by sumoylated transcription factors than [by] their unmodified forms”. But the effect of sumoylation on transcription factors can itself be countered by other regulatory agents — in particular, SUMO proteases, that cleave the bond between SUMO and the transcription factor it has modified. And so, for example, “In the case of the transcription factor ELK1, its sumoylation recruits HDAC2 [histone deacetylase 2], leading to enhanced transcriptional repression. Sumoylated ELK1 is desumoylated mainly by SENP1, and SENP1 depletion dampens transcriptional activation mediated by ELK1” (Hickey, Wilson and Hochstrasser 2012).
- Polycomb Repressor Complex 2 (PRC2) is best known for its histone methylating activity, with a silencing influence upon gene expression. However, PRC2 has now been found also to methylate a transcription factor, GATA4. In one study, this “attenuated [GATA4’s] transcriptional activity by reducing its interaction with and acetylation by p300”. This repression of GATA4-related gene expression is required for normal embryonic development of the heart (He, Shen, Ma et al. 2012).
- One research team, after classifying chromatin into five structural types, found that “DNA binding factors” (DBFs, including transcription factors) bound preferentially to specific chromatin types, despite the fact that the binding motifs were present in all types. “These preferences are most likely due to the presence of chromatin-associated ‘helper’ proteins that assist the DBFs. A helper protein may physically interact with the DBF and stabilize the binding of the DBF to its motifs. ... The chromatin types thus constitute a selection system that guides each DBF to its binding motifs in only some regions of the genome” (Steensel 2011).
- “One mechanism cells use to increase the DNA-binding specifity of TFs is cooperative DNA binding” (Lelli, Slattery and Mann 2012). The authors distinguish three kinds of cooperative binding: (1) classical cooperativity, which “relies on direct protein-protein interactions between TFs and their cofactors to increase DNA-binding affinity”. A variation (“latent specificity”) “is when protein-protein interactions lead not only to increased DNA-binding affinity but also to a change in DNA-binding specificity”. (2) Modular cooperativity, unlike the classical kind, involves TFs not merely in homo- and heterodimers, but in large complexes of proteins. (3) Collaborative competition refers to cooperative binding that “occurs only on a chromatin template because multiple TFs are more effective than are individual TFs at competing with nucleosomes for binding to target sequences”.
- There is also the occurrence of overlapping binding sites. A research team has demonstrated, for example, that three transcription factors bind in varying, mutually exclusive and overlapping combinations at one or both of two DNA promoter binding sites available at many genes. The investigators suggest that “the binding competition between the three factors controls biological processes such as rapid cell growth of both neoplastic and stem cells” (Ngondo-Mbongo, Myslinski, Aster and Philippe Carbon 2013).
- Studies on the glucocorticoid receptor (a transcription factor) show that a TF can respond to, or be affected by, the DNA binding sequence so as to change its own structure. When DNA binding sequences for the glucocorticoid receptor at various genes differ by as little as a single base pair, this difference can (“allosterically”) alter the receptor’s conformation. The regulatory activity of the receptor may therefore change from one gene to another (Meijsing et al. 2009).
- “Nuclear receptors are crucial regulators of gene expression that directly bind to DNA. Now, Maletta et al. describe the structure of ultraspiracle protein/ecdysone receptor (USP/EcR) bound to inverted repeat DNA. Although these inverted repeats of DNA are palindromic [that is, nearly symmetric], upon binding to the receptor the DNA–USP/EcR complex adopts an asymmetrical configuration. This conformational change has functional consequences for the orientation of transcriptional co-activators, such as those required for chromatin remodelling” (note in Nature Reviews Genetics re: Maletta, Orlov, Roblin et al. 2014).
- From an article about a study designed to disentangle the role of the DNA sequence as such from that of the DNA shape in the binding of Hox transcription factors: “[DNA] shape readout is a direct and independent component of binding site selection by Hox proteins” — independent, that is, from the DNA sequence (Abe, Dror, Yang et al. 2015, doi:10.1016/j.cell.2015.02.008). Regarding this study: “The nucleotide sequence alone was a partial predictor of binding specificity, but the predictions were improved by incorporating various inferred shape features of the DNA sequences, such as minor-groove width, roll, propeller twist and helical twist. The modelling also revealed positions in the DNA sequence at which the structural features had a particularly important role in determining which Hox proteins bound” (Burgess 2015, doi:10.1038/nrg3944).
- As an indication of the kind of “remote” event that can yield transcription-factor activity: the intracellular domain (protein fragment) resulting from proteolysis of certain cell surface receptors can migrate through the cytoplasm, binding to various other proteins and thereby affecting cell signaling networks. One result may be the stimulation of transcription factor activity in the nucleus, or the migration of a transcription factor into the nucleus, where it may bind to gene promoters.
- Transcription factors that help to activate gene expression can also play a role in the 3' end processing of the transcripts that are produced — and particularly in polyadenylation (Skinner 2011, citing work by T. Nagaike et al.).
- It is thought today that transcription factors may have other roles beyond direct regulation of transcription. Many transcription factors bind “throughout the genome” at sites that “vastly exceed the number of expected gene targets” and “this raises the intriguing possibility that most binding events of some transcription factors might...have a currently unrecognized role in genome-wide biology” (MacQuarrie, Fong, Morse and Tapscott 2011).
- A new type of transcription factor has recently been found, combining sequence specificity (it binds to a common gene promoter sequence) and nucleosome-like, non-specific DNA binding. Part of the transcription factor contacts DNA “in a manner almost identical to that of core histones within the nucleosome, particularly related to their conserved structural and electrostatic complementarity to DNA”. Moreover, monoubiquitination of a lysine residue in the transcription factor is a prerequisite for monoubiquitination of H2B and methylation of H3 in neighboring, downstream nucleosomes, the latter being preparatory steps for gene activation (Nardini, Gnesutta, Donati et al. 2013).
- Regulatory elements such as transcriptional promoters and enhancers “typically consist of multiple binding sites for transcription factors (TFs) and are often detected and interpreted based on the occurrence of consensus, high-affinity binding motifs for TFs. A new study highlights that, beyond TF binding affinity, the wider genomic context and arrangement of TF binding sites is crucial in tissue-specific enhancers”. For example, for one enhancer “binding sites had low affinity (based on their deviation from consensus sequences); however, their optimal arrangement achieved strong and localized reporter expression in the notochord. Thus, syntax is crucial and can compensate for low-affinity TF binding sites”. In a study of one group of enhancers with two binding sites for one TF and another binding site for a second TF, the activity of the enhancers “is highly dependent on the arrangement and wider context of these binding sites. Further manipulations of the enhancers confirmed key roles for binding site orientation, spacing, and flanking nucleotides” (Burgess 2016, doi:10.1038/nrg.2016.74).
- “In budding yeast: “We observe that maximum promoter activity is determined by TF concentration and not by the number of binding sites. Surprisingly, the addition of an activator site often reduces expression. A thermodynamic model that incorporates competition between neighboring binding sites for a local pool of TF molecules explains this behavior and accurately predicts both absolute expression and the amount by which addition of a site increases or reduces expression. Taken together, our findings support a model in which neighboring binding sites interact competitively when TF is limiting but otherwise act additively” (van Dijk, Sharon, Lotan-Pompan et al. 2016, doi:10.1101/gr.212316.116).
- Regarding the erythroid transcription factor, also known as GATA, and its binding site in an intron portion of a gene relating to a form of anemia: “The first intronic mutations in the intron 1 GATA[-binding] site (int-1-GATA) of 5-aminolevulinate synthase 2 (ALAS2) have been identified in X-linked sideroblastic anemia pedigrees, strongly suggesting [they] could be causal mutations of [this anemia] ... Here, we generated mice lacking a 13 base-pair fragment, including this int-1-GATA site and found that hemizygous deletion led to an embryonic lethal phenotype due to severe anemia resulting from a lack of ALAS2 expression, indicating that this non-coding sequence is indispensable for ALAS2 expression in vivo. Further analyses revealed that this int-1-GATA site anchored the GATA site in intron 8 (int-8-GATA) and the proximal promoter, forming a long-range loop to enhance ALAS2 expression by an enhancer complex including GATA1, TAL1, LMO2, LDB1 and Pol II at least, in erythroid cells” (Zhang, Zhang, An et al. 2017, doi:10.1093/nar/gkw901).
- “Living organisms sense and respond to light, a crucial environmental factor, using photoreceptors, which rely on bound chromophores such as retinal, flavins, or linear tetrapyrroles for light sensing. The discovery of photoreceptors that sense light using 5'-deoxyadenosylcobalamin, a form of vitamin B12 that is best known as an enzyme cofactor, has expanded the number of known photoreceptor families and unveiled a new biological role of this vitamin. The prototype of these B12-dependent photoreceptors, the transcriptional repressor CarH, is widespread in bacteria and mediates light-dependent gene regulation in a photoprotective cellular response. CarH activity as a transcription factor relies on the modulation of its oligomeric state by 5'-deoxyadenosylcobalamin and light” (doi:10.1146/annurev-biochem-061516-044500).
- “Enhancers for embryonic stem (ES) cell-expressed genes and lineage-determining factors are characterized by conventional marks of enhancer activation in ES cells, but it remains unclear whether enhancers destined to regulate cell-type-restricted transcription units might also have distinct signatures in ES cells. Here we show that cell-type-restricted enhancers are ‘premarked’ and activated as transcription units by the binding of one or two ES cell transcription factors, although they do not exhibit traditional enhancer epigenetic marks in ES cells, thus uncovering the initial temporal origins of cell-type-restricted enhancers. This premarking is required for future cell-type-restricted enhancer activity in the differentiated cells, with the strength of the ES cell signature being functionally important for the subsequent robustness of cell-type-restricted enhancer activation” (Kim, Tan, Ma et al. 2018, doi:10.1038/s41586-018-0048-8).
- “Glucocorticoids are potent steroid hormones that regulate immunity and metabolism by activating the transcription factor (TF) activity of glucocorticoid receptor (GR). Previous models have proposed that DNA binding motifs and sites of chromatin accessibility predetermine GR binding and activity. However, there are vast excesses of both features relative to the number of GR binding sites. Thus, these features alone are unlikely to account for the specificity of GR binding and activity ... We found that glucocorticoid treatment induces GR to bind to nearly all pre-established enhancers within minutes. However, GR binds to only a small fraction of the set of accessible sites that lack enhancer marks. Once GR is bound to enhancers, a combination of enhancer motif composition and interactions between enhancers then determines the strength and persistence of GR binding, which consequently correlates with dramatic shifts in enhancer activation. Over the course of several hours, highly coordinated changes in TF binding and histone modification occupancy occur specifically within enhancers, and these changes correlate with changes in the expression of nearby genes. Following GR binding, changes in the binding of other TFs precede changes in chromatin accessibility, suggesting that other TFs are also sensitive to genomic features beyond that of accessibility” (McDowell, Barrera, D’Ippolito et al. 2018, doi:10.1101/gr.233346.117).
-
Additional dynamic aspects of transcription factor activity
A transcription factor’s role in gene expression is often triggered by
external signals. The nature and patterning (in time and space) of the
signal may have a major effect upon the performance of the
transcription factor, and this latter performance may in turn produce
different patterns of expression in the genes — perhaps hundreds of
them — influenced by the transcription factor. In other words, it’s
not just a matter of a straightforward, encoded matching of the
transcription factor with gene promoters, but of the dynamics of the
triggering signal and then the dynamics of the transcription factor’s
interaction with the promoters. All this is only beginning to be
looked at, but the following is suggestive.
- “[Transcription factor] NF-kB, involved in controlling inflammation, undergoes nucleocytoplasmic oscillations in response to tumor necrosis factor-a (TNFa), but sustained nuclear localization in response to bacterial lipopolysaccharides (LPSs). Thus, NF-kB translocation dynamics encode the signal identity (TNFa or LPS). Similarly, the tumor suppressor transcription factor p53 undergoes a dose-dependent number of nuclear pulses in response to DNA breaks, but a single sustained pulse with dose-dependent amplitude and duration in response to ultraviolet irradiation. Thus, p53 dynamics encode both the dose (severity) and the identity of the stress” (Hansen and O’Shea 2013).
- Another example: the yeast transcription factor, Msn2, binds to DNA “stress response elements,” thereby regulating many genes in response to various stresses. “Under normal growth conditions, Msn2 is phosphorylated and localized to the cytoplasm. In the presence of stress stimuli, Msn2 is dephosphorylated, rapidly enters the nucleus and activates gene expression. It is not fully understood how Msn2 is activated by unrelated stresses, nor is it known whether information about stress identity and quantity is conveyed by Msn2 in the process of its activation”. The authors of that statement (Hao and O’Shea 2012) proceed to show how certain dynamic factors help give specificity to the action of this transcription factor.
- In particular, Hao and O’Shea show that “the identities and intensities of different stresses are transmitted by modulation of the amplitude, duration or frequency of nuclear translocation” of Msn2. Distinct dynamical schemes affect particular genes differently. That is, “Different stresses elicit qualitatively different dynamical patterns of transcription factor activation. These patterns are then interpreted by promoters with distinct properties to produce different patterns of target gene expression” (Hao and O’Shea 2012). As for the relevant properties of promoters, see “Promoter activation kinetics” above.
- Reporting on later work by Hao and colleagues on the Msn2 yeast transcription factor: “In the absence of stress, Msn2 is phosphorylated by protein kinase A (PKA) and is located in the cytoplasm, but in response to stress it is dephosphorylated and translocated to the nucleus. The dynamics of Msn2 translocation vary depending on the type of stress, and this variability is thought to result from different oscillating patterns of PKA activity ... Strikingly, the amount of nuclear translocation of Msn2 was highly dependent on the specific dynamics of this PKA inhibition input: high- and low-amplitude oscillations resulted in large and very small amounts of translocation, respectively, whereas a prolonged low-amplitude input resulted in translocation at half the maximum level” (Flintoft 2013).
- More simply, there is the dynamics of competition: when many copies of a given activating transcription factor bind to particular DNA sequences, this can repress genes not associated with these particular sequences. This is thought to occur when the activated genes deplete one or more of the general transcription factors required by the repressed genes.
- A transcription factor can bind to DNA for long periods, or cycle on and off rapidly. The rapid cycling tends to be associated with fast nucleosome turnover rates, as if the transcription factor and the nucleosomes were competing for the same binding sites. These dynamics appear to be more directly related to gene expression than transcription factor “occupancy” of DNA considered without regard to the different dynamic patterns. “We propose that transcription factor binding turnover is a major point of regulation in determining the functional consequences of transcription binding, and is mediated mainly by control of competition between transcription factors and nucleosomes” (Lickwar, Mueller, Hanlon et al. 2012). The authors consider rapid cycling of transcription factors and nucleosomes to be a “poised” transcriptional state. Given the right stimulus, this state can be converted to a stable transcriptional state, mediated perhaps by the eviction of nucleosomes as a result of chromatin remodeling proteins, histone modifications, or replacement of certain histones with histone variants.
- Regarding the dynamics of one particular transcription factor, p53, which affects numerous genes: “Cells that experience p53 pulses recover from DNA damage, whereas cells exposed to sustained p53 signaling frequently undergo senescence. Our results show that protein dynamics can be an important part of a [transcription-regulating] signal, directly influencing cellular fate decisions” (Purvis, Karhohs, Mock et al. 2012).
- “Here, we have examined the binding properties of three Forkhead (FOX) transcription factors, FOXK2, FOXO3 and FOXJ3 in vivo. Extensive overlap in chromatin binding is observed, although underlying differential DNA binding specificity can dictate the recruitment of FOXK2 and FOXJ3 to chromatin. However, functionally, FOXO3-dependent gene regulation is generally mediated not through uniquely bound regions but through regions occupied by both FOXK2 and FOXO3 where both factors play a regulatory role. Our data point to a model whereby FOX transcription factors control gene expression through dynamically binding and generating partial occupancy of the same site rather than mutually exclusive binding derived by stable binding of individual FOX proteins” (Chen, Ji, Webber and Sharrocks 2016, doi:10.1093/nar/gkv1120).
- “The question of how TFs [transcription factors] locate their cognate binding sites (typically spanning over 6-12 nucleotides) scattered over millions to billions of base pairs has remained an enigma”. “Accumulating evidence showing that TF binding sites are embedded within a unique environment, specific to each TF, leads to the hypothesis that the search process is facilitated by favorable DNA features that help to improve the search efficiency”. “We propose that the motif environments that possess favorable features, specific to each TF, may help to narrow down the TF search space, and help to attract the TF to its functional site, thus providing a more efficient search process” (Dror, Rohs and Mandel-Gutfreund 2016, doi:10.1002/bies.201600005).
- “Cofactor squelching is the term used to describe competition between transcription factors (TFs) for a limited amount of cofactors in a cell with the functional consequence that TFs in a given cell interfere with the activity of each other ... recent genome-wide studies have demonstrated that signal-dependent TFs are very often absent from the enhancers that are acutely repressed by those signals, which is consistent with an indirect mechanism of repression such as squelching ... we discuss how TF cooperativity in so-called hotspots and super-enhancers may sensitize these to cofactor squelching”. “We propose that the crosstalk between any two transcriptional activators to a large extent can be described by a combination of cooperativity in cis to synergistically activate shared target genes and by competition in trans to mutually repress non-shared gene programs. Such competition between transcriptional activators may be involved in prioritizing transcription and translation of signal-induced genes over that of cell identify genes, e.g. in response to an inflammatory signal. Conversely, it could also be involved in repression of inflammation by nuclear receptors such as GR, which is activated by the potent anti-inflammatory glucocorticoids. In addition, it is possible that cofactor squelching plays a role during differentiation by indirectly downregulating stem cell genes when specialized gene programs are activated” (Schmidt, Larsen, Loft and Mandrup 2016, doi:10.1002/bies.201600034).
- A group of general regulatory factors (GRFs) in yeast that help to organize chromatin through their interactions with a core consensus DNA sequence were investigated to determine whether their specificity resulted solely from direct base readout. It did not. “We find that computationally predicted DNA shape features (e.g., minor groove width, helix twist, base roll, and propeller twist) that are not defined by a unique consensus sequence are embedded in the nonunique portions of GRF motifs and contribute critically to sequence-specific binding. This dual source specificity occurs at GRF sites in promoter regions where chromatin organization starts. Outside of promoter regions, strong consensus sites lack the shape component and consequently lack an intrinsic ability to bind cognate GRFs, without regard to influences from chromatin. However, sites having a weak consensus and low intrinsic affinity do exist in these regions but are rendered inaccessible in a chromatin environment. Thus, GRF site-specificity is achieved through integration of favorable DNA sequence and shape readouts in promoter regions and by chromatin-based exclusion from fortuitous weak sites within gene bodies” (Rossi, Lai and Pugh 2018, doi:10.1101/gr.229518.117).
-
RNA-binding proteins
“Increasing evidence suggests that transcriptional control and chromatin
activities at large involve regulatory RNAs, which likely enlist specific
RNA-binding proteins ... Multiple RBPs have been implicated in transcription
control ... Like transcription factors, RNA-binding proteins also show
strong preference for hotspots in the genome, particularly gene promoters,
where their association is frequently linked to transcriptional output.
[There is] extensive co-association between transcription factors and
RNA-binding proteins, as exemplified by YY1, a known RNA-dependent
transcription factor, and RBM25, an RNA-binding protein involved in splicing
regulation. Remarkably, RBM25 depletion attenuates all YY1-dependent
activities, including chromatin binding, DNA looping, and transcription. We
propose that various RNA-binding proteins may enhance network interaction
through harnessing regulatory RNAs to control transcription”
(Xiao, Chen, Liang et al. 2019, doi:10.1016/j.cell.2019.06.001).
-
DNA- and RNA-binding proteins
Transcription factors are one of many kinds of DNA-binding proteins.
Throughout this document mention is also made of RNA-binding proteins — for
example, in connection with mRNA splicing. However, attention is now being
given also to proteins that bind both DNA and RNA. Because these
proteins can regulate gene expression at multiple levels, they could be
treated in more than one section of this document. For details, see
Proteins that bind both DNA and RNA under
POST-TRANSCRIPTIONAL DECISION-MAKING
below.
-
CpG islands
CpG islands are rich in the genomic nucleotide bases (“letters”) G and C,
and more particularly in CpG dinucleotides (that is, adjacent CGs — not
base-paired, but rather as neighbors along the length of one strand, with
the C toward the 5' end of the strand and the G toward the 3' end). These
islands are often associated with gene promoters, where they tend not to be
methylated (unlike CpG dinucleotides scattered throughout the rest of DNA,
outside islands). However, “most, perhaps all, CpG islands are sites of
transcription initiation” even though many of them are not associated with
currently recognized promoters (Deaton and Bird 2011).
“CpG islands (CGIs) represent a widespread feature of vertebrate genomes,
being associated with ~70% of all gene promoters. CGIs control transcription
initiation by conferring nearby promoters with unique chromatin properties.
In addition, there are thousands of distal or orphan CGIs (oCGIs) whose
functional relevance is barely known. Here we show that oCGIs are an
essential component of poised enhancers that augment their long-range
regulatory activity and control the responsiveness of their target genes ...
oCGIs act as tethering elements that promote the physical and functional
communication between poised enhancers and distally located genes,
particularly those with large CGI clusters in their promoters. Therefore, by
acting as genetic determinants of gene–enhancer compatibility, CGIs can
contribute to gene expression control under both physiological and
potentially pathological conditions” (Pachano, Sánchez-Gaya, Ealo et al.
2021, doi:10.1038/s41588-021-00888-x).
- The different ways in which transcription factors and chromatin remodeling/restructuring factors interact with CpG islands (which have their own subtle variations in structure) play a large role in gene regulation in vertebrates.
- Particularly in embryonic stem cells, some protein complexes “attracted” by CpG islands apply gene-activating marks to the nearby chromatin, while other protein complexes apply gene-repressing marks. This is apparently associated with the tendency for many genes in stem cells to be held in a bivalent or “poised” state, ready to be quickly activated or repressed depending on developmental requirements (Deaton and Bird 2011).
- DNA sequence-specific transcriptional regulators play an important role in swinging the bivalent state toward a more definite activating or repressive condition (Deaton and Bird 2011).
- Imprinted genes are often associated with repressive methylation of a CpG island in a regulatory locus.
- See DNA methylation below.
-
Co-activators and co-repressors
Co-activators and co-repressors (they are generally proteins) do not bind
directly to DNA regulatory sequences, but are typically recruited by
transcription factors.
“Pleiotropically acting eukaryotic corepressors such as retinoblastoma and
SIN3 have been found to physically interact with many widely expressed
‘housekeeping’ genes. Evidence suggests that their roles at these loci are
not to provide binary on/off switches, as is observed at many highly
cell‐type specific genes, but rather to serve as governors, directly
modulating expression within certain bounds, while not shutting down gene
expression. This sort of regulation is challenging to study, as the
differential expression levels can be small. We hypothesize that depending
on context, corepressors mediate ‘soft repression,’ attenuating expression
in a less dramatic but physiologically appropriate manner. Emerging data
indicate that such regulation is a pervasive characteristic of most
eukaryotic systems, and may reflect the mechanistic differences between
repressor action at promoter and enhancer locations. Soft repression may
represent an essential component of the cybernetic systems underlying
metabolic adaptations, enabling modest but critical adjustments on a
continual basis”
(Mitra, Raicu, Hickey et al. 2021, doi:10.1002/bies.202000231)
“So far, the mild transcriptional regulation of housekeeping genes by the Rb
or the Sin3 complex has been mostly overlooked or dismissed as off‐target
effects. Expression profile analysis of these housekeeping genes points to
significant yet understudied roles in development. The authors propose that
these corepressor complexes fine‐tune the expression of these genes
throughout development. In doing so, they contribute to an essential
transcriptional control for proper development”
(Plessel and David 2021, doi:10.1002/bies.202000326).
- Co-activators and co-repressors often play a role in modifying histone tails, repositioning or removing nucleosomes, and restructuring chromatin.
- Co-activators can also alter the DNA-binding specificity of the transcription factors they associate with (see the original research cited in Stower 2012).
- As for co-repressors, the emerging concept is that “long-term gene repression is probably maintained not by the constitutive presence of co-repressor complexes but by histone modifications that are maintained by intermittent co-repressor activity. Current models of co-repressor function appreciate the dynamics of the opposing co-activator and co-repressor complexes, which seem to continually cycle on and off DNA. As genome-wide data continue to accrue, co-repressor complexes may turn out to be as important in gene-activation events as in repression owing to, for example, their ability to reset chromatin for subsequent rounds of transcription” (Perissi 2010).
- New techniques are making it possible to assess more fully how various co-factors cooperate with the transcription factors binding to DNA. In one case more than 40 co-activators were found tethered to a transcription factor on a gene enhancer. These included Mediator, which links directly to the transcription complex, and other large complexes that loosen the chromatin structure to facilitate transcription (Sela, Chen, Martin-Brown et al. 2012). “Researchers know that all DNA-binding factors partner with other proteins to switch genes on or off. What is remarkable here is their sheer number. ‘It would be very interesting to find out whether this is the norm’, says [research team leader] Ron Conaway” (Physorg 2012b).
- “Many proteins originally identified as cytoplasmic — including many associated with the cytoskeleton or cell junctions — are increasingly being found in the nucleus, where they have specific functions. Here, we focus on proteins that translocate from the cytoplasm to the nucleus in response to external signals and regulate transcription without binding to DNA directly (for example, through interaction with transcription factors). We propose that proteins with such characteristics are classified as a distinct group of extracellular signalling effectors, and [their roles include] linking cell morphology and adhesion with changes in transcriptional programmes in response to signals such as mechanical stresses” (Lu, Muers and Lu 2016, doi:10.1038/nrm.2016.41).
- A single co-repressor example: HDAC3 (histone deacetylase 3). “HDAC3 is unique among the HDAC family proteins as the major HDAC that functions as part of nuclear receptor co-repressor complexes and requires them for its catalytic activity. HDAC3 is vital to life because it regulates many developmental, physiological and metabolic processes. Throughout development, HDAC3 serves to integrate complex signals from the environment and deliver them onto the genome to influence cellular migration, differentiation, fate and growth. In adult mammals, HDAC3 uniquely regulates major metabolic and energy utilization pathways and circadian, nutrient and environmental challenges, influences memory formation, limits autoimmunity and protects skeletal integrity, among other homeostatic functions ... [Research has revealed] an extraordinarily diverse set of tissue-specific HDAC3 targets [and] a remarkably diverse set of nuclear receptors and other transcription factors that recruit HDAC3 to chromatin, histone and non-histone targets and to participate in enzymatic and non-enzymatic functions” (Emmett and Lazar 2019, doi:10.1038/s41580-018-0076-0).
-
From foregoing article by Emmett and Lazar:
- “HDac3 is a core component of nuclear receptor co-repressor complexes that modulate nuclear receptor-mediated transcription.”
- “HDAC3 controls lipid metabolism and circadian histone deacetylation in the liver.”
- “HDac3 suppresses liver metabolism and circadian clock genes through distinct enhancer complexes.”
- “HDAC3 primes the thermogenic capacity of brown fat for survival in cold conditions.”
- “HDAC3 influences cardiac development and cardiomyocyte metabolism.”
- “HDAC3 regulates neuronal cell fate and function.”
- “HDAC3 coordinates lung development.”
- “HDac3 controls brain development, glial cell fate and the formation of long-term memory.”
- “HDAC3 governs intestinal homeostasis and host defence.”
- “HDAC3 in pancreatic β-cells controls glucose-stimulated insulin secretion.”
- “HDAC3 impacts skeletal muscle metabolism and a fuel source switch.”
- “HDAC3 promotes haematopoietic cell lineages and prevents a lethal autoimmune disease.”
- “HDAC3 is required for the development and physiological remodelling of bones.”
- “During the activation of mouse macrophages by lipopolysaccharides, histone deacetylase 3 controls inflammatory responses by both repressing and activating gene transcription depending on its differential association with transcription factors” (Nguyen, Adlanmerini, Hauck and Lazar 2020, doi:10.1038/s41586-020-2576-2).
-
Enhancers and silencers
Enhancers are DNA sequences that play a role in activating target genes,
often from a distance, and do so, especially during development, in a time-
and tissue-dependent way. They feature both in disease and development,
and act at least in part by means of chromosome looping. This is thought
to be facilitated by the binding of transcription factors to both the
enhancer and promoter sites. Silencers are similar, except that they help
to silence or repress gene expression. However, the distinction between
enhancers and silencers may turn out not to be so clear-cut — and, indeed,
the distinction between enhancers and promoters may not be so clear-cut
(see below).
“One of the main questions that needs to be addressed is at which step
during gene activation do various nucleoprotein complexes assemble at
distant enhancers, and how do these complexes then contribute to promoter
accessibility, PIC recruitment and/or assembly, and transcription initiation
and elongation? Enhancers have been shown to have a role in: PIC recruitment
at target promoters, removing proteasome complexes at promoters, the
generation of intrachromosomal loops between regulatory regions, and the
regulation of elongation. Enhancers are also involved in the removal of
repressive histone modifications, suggesting that they also contribute to
the delivery of enzymes that regulate histone modifications”
(Vernimmen and Bickmore 2015, doi:10.1016/j.tig.2015.10.004).
Studies “suggest that there may be many common mechanisms involved in
enhancer–promoter communication ...
- Enhancers are first primed by pioneer transcription factors (TFs).
- Other TFs are likely required for subsequent events.
- There is a hierarchy between enhancers and the promoters that they regulate.
- Enhancers and promoters share similar properties, but differ in the characteristics and the abundance of the RNAs that they produce.
- By recruiting the preinitiation complex and other proteins, enhancers have a role of increasing the concentration of the transcription machinery at target promoters” (Vernimmen and Bickmore 2015, doi:10.1016/j.tig.2015.10.004).
“The exact composition of core promoter elements may be a key determinant of
enhancer-promoter specificity. In mammalian genomes, enhancers are enriched
in core promoter elements but are CpG poor, whereas promoters are generally
CpG rich. Beside the CpG content, enhancers and promoters have broad
similarities and overlapping functional properties, and have been considered
to form a single class of regulatory element” (Vernimmen and Bickmore 2015,
doi:10.1016/j.tig.2015.10.004).
“Distant-acting tissue-specific enhancers, which regulate gene expression,
vastly outnumber protein-coding genes in mammalian genomes ... [Single and
combinatorial enhancer deletions at seven distinct mouse loci required for
limb development revealed, unexpectedly, that] none of the ten deletions of
individual enhancers caused noticeable changes in limb morphology. By
contrast, the removal of pairs of limb enhancers near the same gene resulted
in discernible phenotypes, indicating that enhancers function redundantly in
establishing normal morphology. [Tests suggested that] functional
redundancy is conferred by additive effects of enhancers on gene expression
levels. A genome-wide analysis integrating epigenomic and transcriptomic
data from 29 developmental mouse tissues revealed that mammalian genes are
very commonly associated with multiple enhancers that have similar
spatiotemporal activity. Systematic exploration of three representative
developmental structures (limb, brain and heart) uncovered more than one
thousand cases in which five or more enhancers with redundant activity
patterns were found near the same gene. Together, our data indicate that
enhancer redundancy is a remarkably widespread feature of mammalian genomes
that provides an effective regulatory buffer to prevent deleterious
phenotypic consequences upon the loss of individual enhancers”
(Osterwalder, Barozzi, Tissières et al. 2018, doi:10.1038/nature25461).
Some key recent developments, according to Rickels and Shilatifard 2018
(doi:10.1016/j.tcb.2018.04.003, directly quoted):
• Metazoan development requires the orchestration of hundreds of thousands of enhancers to establish precise spatiotemporal gene expression patterns.
• Enhancers commonly exist in a ‘suboptimal’ state with respect to their transcription factor binding affinities, and this evolutionary ‘suboptimization’ of both the sequence and binding motif arrangement is key to encoding enhancer tissue-specificity.
• Accumulating evidence suggests that enhancers regulate gene transcription by stimulating release of promoter-paused RNA polymerase II into productive elongation.
• Bidirectional transcription of enhancer DNA is now appreciated to be a general characteristic of active enhancers, and recent reports document numerous examples of how promoters can function as enhancers to stimulate long-range gene activation. Thus, the distinction between enhancers and promoters is becoming less apparent.
• Clusters of cis-regulatory elements appear to be highly interconnected in the nucleus, and these complex regulatory ‘hubs’ are organized into topological domains along the linear chromosome.
“The current paradigm in the field of gene regulation postulates that
regulatory information for generating gene expression is organized into
modules (enhancers), each containing the information for driving gene
expression in a single spatiotemporal context. This modular organization is
thought to facilitate the evolution of gene expression by minimizing
pleiotropic effects. Here we review recent studies that provide evidence of
quite the opposite: (i) enhancers can function in multiple developmental
contexts, implying that enhancers can be pleiotropic, (ii) transcription
factor binding sites within pleiotropic enhancers are reused in different
contexts, and (iii) pleiotropy impacts the structure and evolution of
enhancers. Altogether, this evidence suggests that enhancer pleiotropy is
pervasive in animal genomes, challenging the commonly held view of
modularity” (Sabarís et al. 2019, doi:10.1016/j.tig.2019.03.006).
“While active enhancers are characterized by open chromatin structure, not
all open enhancers are active. The binary distinction into open/closed
regions and active/inactive enhancers is insufficient to describe this
complex relationship. Instead, considering quantitative differences in the
accessibility signal allows for discriminating between different regulatory
states” (table of contents blurb for Bozek and Gompel 2020,
doi:10.1002/bies.201900188).
“Shadow enhancers are seemingly redundant transcriptional cis-regulatory
elements that regulate the same gene and drive overlapping expression
patterns. Recent studies have shown that shadow enhancers are remarkably
abundant and control most developmental gene expression in both
invertebrates and vertebrates, including mammals. Shadow enhancers might
provide an important mechanism for buffering gene expression against
mutations in non-coding regulatory regions of genes implicated in human
disease. Technological advances in genome editing and live imaging have shed
light on how shadow enhancers establish precise gene expression patterns and
confer phenotypic robustness. Shadow enhancers can interact in complex ways
and may also help to drive the formation of transcriptional hubs within the
nucleus. Despite their apparent redundancy, the prevalence and evolutionary
conservation of shadow enhancers underscore their key role in emerging
metazoan gene regulatory networks”
(Kvon, Waymack, Gad and Wunderlich 2021, doi:10.1038/s41576-020-00311-x).
“Silencers are regulatory DNA elements that reduce transcription from their
target promoters; they are the repressive counterparts of enhancers.
Although discovered decades ago, and despite evidence of their importance in
development and disease, silencers have been much less studied than
enhancers. Recently, however, a series of papers have reported systematic
studies of silencers in various model systems. Silencers are often
bifunctional regulatory elements that can also act as enhancers, depending
on cellular context, and are enriched for expression quantitative trait loci
(eQTLs) and disease-associated variants. There is not yet evidence of a
‘silencer chromatin signature’, in the distribution of histone modifications
or associated proteins, that is common to all silencers; instead, silencers
may fall into various subclasses, acting by distinct (and possibly
overlapping) mechanisms”
(Segert, Gisselbrecht and Bulyk 2021, doi:10.1016/j.tig.2021.02.002).
“Enhancers are central to control ... tissue-specific gene expression
pattern ... We find that enhancers showing tissue-specific activity are
highly enriched in intronic regions and regulate the expression of genes
involved in tissue-specific functions, whereas housekeeping genes are more
often controlled by intergenic enhancers, common to many tissues. Notably,
an intergenic-to-intronic active enhancers continuum is observed in the
transition from developmental to adult stages: the most differentiated
tissues present higher rates of intronic enhancers, whereas the lowest rates
are observed in embryonic stem cells. Altogether, our results suggest that
the genomic location of active enhancers is key for the tissue-specific
control of gene expression”
(Borsari, Villegas-Mirón, Pérez-Lluch et al. 2021, doi:10.1101/gr.270371.120)
“Dual-function regulatory elements (REs), acting as enhancers in some
cellular contexts and as silencers in others ... We herein investigated this
class of REs in the human genome and profiled their activity across multiple
cell types. Focusing on enhancer–silencer transitions specific to the
development of T cells, we built an accurate deep learning classifier of REs
and identified about 12,000 silencers active in primary peripheral blood T
cells that act as enhancers in embryonic stem cells. Compared with regular
silencers, these dual-function REs are evolving under stronger purifying
selection and are enriched for mutations associated with disease phenotypes
and altered gene expression. In addition, they are enriched in the loci of
transcriptional regulators, such as transcription factors (TFs) and
chromatin remodeling genes. Dual-function REs consist of two intertwined but
largely distinct sets of binding sites bound by either activating or
repressing TFs, depending on the type of RE function in a given cell line.
This indicates the recruitment of different TFs for different regulatory
modes and a complex DNA sequence composition of these REs with dual
activating and repressive encoding. With an estimated >6% of cell
type–specific human silencers acting as dual-function REs, this overlooked
class of REs requires a specific investigation on how their inherent
functional plasticity might be a contributing factor to human diseases”
(Huang and Ovcharenko 2022, doi:10.1101/gr.275992.121).
“We identify simple rules for enhancer-promoter compatibility: most
enhancers activated all promoters by similar amounts, and intrinsic enhancer
and promoter activities combine multiplicatively to determine RNA output. In
addition, two classes of enhancers and promoters showed subtle preferential
effects. Promoters of housekeeping genes contained built-in activating
motifs for factors such as GABPA and YY1, which decreased the responsiveness
of promoters to distal enhancers. Promoters of variably expressed genes
lacked these motifs and showed stronger responsiveness to enhancers.
Together, this systematic assessment of enhancer-promoter compatibility
suggests a multiplicative model tuned by enhancer and promoter class to
control gene transcription in the human genome”
(Bergman, Jones, Liu et al. 2022, doi:10.1038/s41586-022-04877-w).
“Paralogues (divergent duplicated genes) are often involved in the same
developmental and cellular processes. In Drosophila, these genes are
frequently separated by large genomic distances. Levo et al. used
quantitative single-cell live imaging to analyse the transcriptional
dynamics of such genes, to determine whether they are co-regulated. The team
identified ‘topological operons’, identifying co-regulation by shared
enhancers and co-transcriptional initiation over distances of nearly 250 kb.
The coordinated transcriptional dynamics arise from associations with
discrete promoter-proximal tethering elements that enable contacts between
these genes in 3D throughout the fly genome”
(Koch 2022, doi:10.1038/s41576-022-00502-8).
“We find that the ultralong distance enhancer network has a nested
multilayer architecture that confers functional robustness of gene
expression. Experimental characterization reveals that enhancer epistasis is
maintained by three-dimensional chromosomal interactions and BRD4
condensation”
(Lin, Liu, Liu et al. 2022, doi:10.1126/science.abk3512).
“Housekeeping genes are considered to be regulated by common enhancers
across different tissues. Here we report that most of the commonly expressed
mouse or human genes across different cell types, including more than half
of the previously identified housekeeping genes, are associated with cell
type–specific enhancers. Furthermore, the binding of most transcription
factors (TFs) is cell type–specific. We reason that these cell type
specificities are causally related to the collective TF recruitment at
regulatory sites, as TFs tend to bind to regions associated with many other
TFs and each cell type has a unique repertoire of expressed TFs. Based on
binding profiles of hundreds of TFs from HepG2, K562, and GM12878 cells, we
show that 80% of all TF peaks overlapping H3K27ac signals are in the top
20,000–23,000 most TF-enriched H3K27ac peak regions, and approximately
12,000–15,000 of these peaks are enhancers (nonpromoters). Those enhancers
are mainly cell type–specific and include those linked to the majority of
commonly expressed genes. Moreover, we show that the top 15,000 most
TF-enriched regulatory sites in HepG2 cells, associated with about 200 TFs,
can be predicted largely from the binding profile of as few as 30 TFs.
Through motif analysis, we show that major enhancers harbor diverse and
clustered motifs from a combination of available TFs uniquely present in
each cell type. We propose a mechanism that explains how the highly focused
TF binding at regulatory sites results in cell type specificity of enhancers
for housekeeping and commonly expressed genes”
(Zhu and Landsman 2023; doi:10.1101/gr.278130.123).
- “An extensive study of [enhancer] logic in the sea urchin Endo16 gene, identified 55 binding sites for 16 regulatory proteins, which form an intricate regulatory computer spanning 2300 base pairs of DNA” (Swanson, Evans and Barolo 2010, citing work by E. H. Davidson).
- Some enhancers have been reported to act as silencers in special circumstances, depending on how they are bound by transcription factors.
- Various histone modifications (“marks” — for example, H3K4me1, H3K4me3, H3K27ac, and H3K36me3) as well as post-translational phosphorylation of RNA polymerase II combine in different ways to influence the status of any given enhancer — highly active, less active, and poised, with two subclasses of poised enhancers. It has been proposed that not only the particular combination of marks, but also their quantitative levels, bear on gene expression, with the latter playing a role in fine-tuning. “We hypothesize that our findings, in which only a fraction of all possible histone modifications were investigated, represent the ‘tip of the iceberg’ with respect to functional refinement of gene enhancer elements” (Zentner, Tesar and Scacheri 2011).
- Research on mouse erythroid cells has suggested that “intragenic enhancers [enhancers located within genes, and whose function as enhancers may relate to genes other than the one in which they are located] behave like alternative erythroid-specific promoters” for the genes in which they are located. That is, they can function much like canonical transcription start sites, from which alternatively spliced mRNAs as well as a variety of short, bidirectional, non-polyadenylated transcripts, are produced. Even when the regular promoter of the relevant gene is deleted, the enhancers account for 50% of the mRNAs produced from it (Casci 2012, reporting on Kowalczyk, Hughes, Garrick et al. 2012).
- In a research paper “the authors suggest that transcription at intragenic enhancers interferes with and attenuates host gene expression, but that whether this, overall, results in the attenuation, fine-tuning or activation of gene expression depends on the balance between enhancer-mediated gene activation and enhancer-mediated interference at each host gene” (Wrighton 2017, doi:10.1038/nrg.2017.90).
- Studies using mouse erythroid cells suggest that tissue-specific enhancers can act at long range to remove both the repressive Polycomb complex and (by recruiting an expression-activating histone demethylase) the H3K27me3 histone mark from developmentally regulated genes (Vernimmen, Lynch, De Gobbi et al. (2011).
- While noncoding DNA is usually the place where one looks for enhancers, a substantial number of enhancers have now been identified as overlapping exons of protein-coding genes. One of these was shown to interact with the promoter of a gene 900 kilobases away. “These results demonstrate that DNA sequences can have a dual function, operating as coding exons in one tissue and enhancers of nearby gene(s) in another tissue, suggesting that phenotypes resulting from coding mutations could be caused not only by protein alteration but also by disrupting the regulation of another gene” (Birnbaum, Clowney, Agamy et al. 2012).
- “Studies have revealed that in different cell types, the repertoire of specific enhancers provides a unique context for the activation of different transcriptional programs in response to signal-dependent transcription factors ... Here, our results further suggest that targets of cell-specific enhancers are already hardwired into the chromatin architecture in each cell lineage. We therefore propose that cell-type-specific looping structure, by controlling the accessibility of the enhancers to their specific [promoter] targets, may form an additional layer of regulation in determining the distinct transcription programs in different cell types” (Jin, Li, Dixon et al. 2013, doi:10.1038/nature12644).
- “Enhancers lie at the nexus of transcription, nuclear organization, chromatin structure, epigenetics, and noncoding RNA. In accordance with such a complex spectrum of biological functions, it seems unlikely that enhancers constitute a monolithic class of regulatory element that works via a single, unified mechanism” (Bulger and Groudine 2011).
- “Transcriptional bursts are believed to be a general property of gene expression. They involve multiple consecutive RNA polymerase complexes being released from promoters to rapidly produce several transcripts, followed by a period of little activity. A new study uses live-imaging techniques to monitor transcriptional bursts in Drosophila melanogaster embryos during development and shows that enhancers can dynamically and coordinately regulate burst frequencies at multiple promoters ... The investigators observed that different enhancers drive different bursting frequencies but have similar burst sizes (that is, a similar number of transcripts per burst). This suggests that burst frequency is a key parameter in controlling gene activity. [Evidence indicates] that the enhancer can activate separate promoters simultaneously” (Cloney 2016, doi:10.1038/nrg.2016.81).
- Super-enhancers. “The ESC [embryonic stem cell] master transcription factors form unusual enhancer domains at most genes that control the pluripotent state. These domains, which we call super-enhancers, consist of clusters of enhancers that are densely occupied by the master regulators and [the pre-initiation complex protein,] Mediator. Super-enhancers differ from typical enhancers in size, transcription factor density and content, ability to activate transcription, and sensitivity to perturbation. Reduced levels of Oct4 or Mediator cause preferential loss of expression of super-enhancer-associated genes relative to other genes, suggesting how changes in gene expression programs might be accomplished during development. In other more differentiated cells, super-enhancers containing cell-type-specific master transcription factors are also found at genes that define cell identity. Super-enhancers thus play key roles in the control of mammalian cell identity” (Whyte, Orlando Hnisz et al. 2013).
- Super-enhancers. “Here, we report that super-enhancers drive the biogenesis of master miRNAs crucial for cell identity by enhancing both transcription and Drosha/DGCR8-mediated primary miRNA (pri-miRNA) processing. Super-enhancers, together with broad H3K4me3 domains, shape a tissue-specific and evolutionarily conserved atlas of miRNA expression and function. CRISPR/Cas9 genomics revealed that super-enhancer constituents act cooperatively and facilitate Drosha/DGCR8 recruitment and pri-miRNA processing to boost cell-specific miRNA production. The BET-bromodomain inhibitor JQ1 preferentially inhibits super-enhancer-directed cotranscriptional pri-miRNA processing. Furthermore, super-enhancers are characterized by pervasive interaction with DGCR8/Drosha and DGCR8/Drosha-regulated mRNA stability control, suggesting unique RNA regulation at super-enhancers. Finally, super-enhancers mark multiple miRNAs associated with cancer hallmarks. This study presents ... an unrecognized higher-order property of super-enhancers in RNA processing beyond transcription” (Suzuki, Young, and Sharp 2017, doi:10.1016/j.cell.2017.02.015).
- Super-enhancers. “It is proposed that the multiple enhancer elements associated with locus control regions and super‐enhancers recruit RNA polymerase II and efficiently assemble elongation competent transcription complexes that are transferred to target genes by transcription termination and transient looping mechanisms. It is well established that transcription complexes are recruited not only to promoters but also to enhancers, where they generate enhancer RNAs. Transcription at enhancers is unstable and frequently aborted. Furthermore, the Integrator and WD‐domain containing protein 82 mediate transcription termination at enhancers. Abortion and termination of transcription at the multiple enhancers of locus control regions and super‐enhancers provide a large pool of elongation competent transcription complexes. These are efficiently captured by strong basal promoter elements at target genes during transient looping interactions” (table of contents blurb for Gurumurthy, Shen, Gunn and Bungert 2018, doi:10.1002/bies.201800164).
-
When genome-wide association studies (GWAS) identify disease risk
variants, the challenge is to find the relevant genes associated with the
risk. Sometimes it is merely assumed that the gene nearest to the
variant is probably the relevant one. However, by mapping the loops
formed by enhancer-promoter interactions in rare, disease-relevant cell
types, researchers have come up with a vastly more complex story. “The
authors identified over 10,000 chromatin loops that were shared by all
three cell types. Importantly, 91% of the loop anchors were associated
with either an enhancer or a promoter, with a median distance of 130 kb
between the anchors ... the authors found that the disease-associated
variants interacted with from zero to ten target genes. For the 684
autoimmune disease–associated variants analyzed, there were 2,597 target
genes mapped through the chromatin interactions. Critically, only 14% of
target genes were the nearest gene to the disease-associated variant, 86%
of variants skipped at least one gene before reaching the target gene and
64% of variants connected to more than one gene
In illustrating how to track down functional information relating to the risk variants, the researchers report that “the risk allele of the rs1537373 variant showed increased interaction with the CDKN2A promoter and the enhancer in the long noncoding RNA (lncRNA) ANRIL. This is a terrific example of what the field is up against, as not only will disease-associated variants synergistically act on multiple genes, but there may also be more complex gene-regulatory mechanisms involved, like ones affecting the function of noncoding RNAs”
A relevant side note: “Could the same information be recovered from linear 1D data? ... the findings emphasize that, even in highly related cell types, a proportion of enhancer-interacting signals will only be captured in 3D”.
(Trynka 2017, doi:10.1038/ng.3982). - “The number of predicted enhancers is about tenfold the number of genes; it remains unclear whether this represents regulation of gene expression in an additive and/or in a redundant manner. Osterwalder et al. used the mouse developing limb to study enhancer function during morphogenesis. Individually deleting ten conserved enhancers of genes associated with mouse and human congenital limb malformation caused no significant change in target-gene expression and, importantly, no limb abnormalities. This indicated that many conserved limb enhancers are not individually essential for limb morphogenesis. The selected panel of enhancers included three enhancer pairs with overlapping limb activity and the same predicted target gene. In two out of three cases, embryos with homozygous deletions of the enhancer pair showed reduction in target-gene expression and limb abnormalities” (Zlotorynski 2018, doi:10.1038/nrm.2018.15).
- Enhancer networks: “Our analyses show extensive correlated activity among enhancers and reveal clusters of enhancers whose activities are coordinately regulated by multiple potential mechanisms involving shared transcription factor binding, chromatin modifying enzymes and 3D chromatin structure, which ultimately co-regulate functionally linked genes” (Malin, Aniba and Hannenhalli 2013).
-
Exons and introns
“Several new studies indicate that rapidly cycling cells constrain
gene-architecture toward short genes with a few introns, allowing efficient
expression during short cell cycles. In contrast, longer genes with long
introns exhibit delayed expression, which can serve as timing mechanisms
for patterning processes [such as occur during embryonic development].
These findings indicate that cell cycle constraints drive the evolution of
gene-architecture and shape the transcriptome of a given cell type”
(Heyn, Kalinka, Tomancak and Neugebauer 2015, doi:10.1002/bies.201400138).
“Introns are ubiquitous features of all eukaryotic cells. Introns need to be
removed from nascent messenger RNA through the process of splicing to
produce functional proteins. Here we show that the physical presence of
introns in the genome promotes cell survival under starvation conditions. A
systematic deletion set of all known introns in budding yeast genes
indicates that, in most cases, cells with an intron deletion are impaired
when nutrients are depleted. This effect of introns on growth is not linked
to the expression of the host gene, and was reproduced even when translation
of the host mRNA was blocked. Transcriptomic and genetic analyses indicate
that introns promote resistance to starvation by enhancing the repression of
ribosomal protein genes that are downstream of the nutrient-sensing TORC1
and PKA pathways. Our results reveal functions of introns that may help to
explain their evolutionary preservation in genes, and uncover regulatory
mechanisms of cell adaptations to starvation”
(Parenteau, Maignon, Berthoumieux et al. 2019,
doi:10.1038/s41586-018-0859-7).
“Introns regulate all levels of their host gene expression, including
transcription, export, RNA stability, and even translation through the exon
junction complex. Introns can also act beyond their host genes”
(Parenteau and Elela 2019, doi:10.1016/j.tig.2019.09.010).
“Molecular signatures defining quiescence in muscle satellite cells (mSCs)
remain enigmatic ... Yue et al. adapted an in vivo fixation approach to
isolate dormant mSCs from healthy muscle. Characterizing the transcriptome
from these cells, they identified intron retention as a novel hallmark of
mSC quiescence”
(Nakka, Kovac, Wong and Dilworth 2020, doi:10.1016/j.devcel.2020.05.028).
“Intragenic regions that are removed during maturation of the RNA
transcript—introns — are universally present in the nuclear genomes of
eukaryotes. The budding yeast, an otherwise intron-poor species, preserves
two sets of ribosomal protein genes that differ primarily in their introns
... We show that introns can mediate inducible phenotypic heterogeneity that
confers a clear fitness advantage. Osmotic stress leads to bimodal
expression of the small ribosomal subunit protein Rps22B, which is mediated
by an intron in the 5′ untranslated region of its transcript. The two
resulting yeast subpopulations differ in their ability to cope with
starvation. Low levels of Rps22B protein result in prolonged survival under
sustained starvation, whereas high levels of Rps22B enable cells to grow
faster after transient starvation. Furthermore, yeasts growing at high
concentrations of sugar, similar to those in ripe grapes, exhibit bimodal
expression of Rps22B when approaching the stationary phase. Differential
intron-mediated regulation of ribosomal protein genes thus provides a way to
diversify the population when starvation threatens in natural environments.
Our findings reveal a role for introns in inducing phenotypic heterogeneity
in changing environments, and suggest that duplicated ribosomal protein
genes in yeast contribute to resolving the evolutionary conflict between
precise expression control and environmental responsiveness”
(Lukačišin, Espinosa-Cantú and Tobias Bollenbach 2022,
doi:10.1038/s41586-022-04633-0).
-
Insulators
An insulator is a boundary sequence in the genome that can play various
roles — and the list of roles is now expanding.
“Insulators play a critical role in spatiotemporal gene regulation in
animals. The evolutionarily conserved CCCTC-binding factor (CTCF) is
required for insulator function in mammals, but not all of its binding sites
act as insulators ... We find that insulation potency depends on the number
of CTCF-binding sites in tandem. Furthermore, CTCF-mediated insulation is
dependent on upstream flanking sequences at its binding sites. CTCF-binding
sites at topologically associating domain boundaries are more likely to
function as insulators than those outside topologically associating domain
boundaries, independently of binding strength. We demonstrate that
insulators form local chromatin domain boundaries and weaken
enhancer–promoter contacts. Taken together, our results provide genetic,
molecular and structural evidence connecting chromatin topology to the
action of insulators in the mammalian genome”
(Huang, Zhu, Jussila et al. 2021, doi:10.1038/s41588-021-00863-6).
-
When bound by various proteins that recognize the particular
insulator sequence, the insulator may serve to:
- block the activity of enhancers (when the insulator is located between an enhancer and the gene regulated by the enhancer);
- serve as an attachment point for the configuration of a chromosome loop, which may have both gene activating and gene repressing effects;
- prevent the further spread of chromatin past the insulator site. [However, “Insulators do not appear to be necessary to prevent the spread of heterochromatin, as proposed previously” (Van Bortle and Corces 2012).]
- “Recent information suggests that their function is more nuanced and depends on the nature of the sequences brought together by contacts between specific insulator sites” (Yang and Corces 2012).
- “In addition to blocking enhancer–promoter interactions, insulators can also direct enhancers to the appropriate promoters. Insulators can not only block the spreading of heterochromatin but they can also demarcate the boundaries between a variety of epigenetic states. Furthermore, the effect of insulators on genome biology goes beyond their involvement in transcription processes as they are also involved in regulating V(D)J recombination [a type of genetic recombination that occurs on a large scale in the immune system]” (Yang and Corces 2012).
- “Insulator proteins also localize to transcription factories, which suggests a role in directing the localization of target genes to nuclear substructures for regulation”. Further, “Transcription factors and insulator-independent complexes also contribute to the organization of coregulated genes for coordinated expression, which suggests that nuclear organization and appropriate genome function depend on numerous additional factors” (Van Bortle and Corces 2012).
- “Preliminary findings sugest that insulators can collaborate, perhaps to establish robust complexes capable of facilitating stable long-range interactions. Insulators also appear to be developmentally regulated by recruitment of both DNA-binding insulator proteins and additional cofactors” (Van Bortle and Corces 2012).
- In humans, tDNAs — DNA sequences transcribed into transfer RNAs (tRNAs) — can act as insulators. Apparently the chromatin environment amenable to tRNA transcription by RNA polymerase III can insulate neighboring protein-coding genes from the elements required for RNA polymerase II transcription. tDNAs may also “have inherent molecular properties that allow them to be suborned as insulators” (Raab, Chiu, Zhu et al. 2012).
- The CTCF protein (which recruits cohesin) plays a central role in insulator function. See “Insulator protein CTCF” under THREE-DIMENSIONAL ORGANIZATION OF CHROMOSOMES, NUCLEUS, AND CELL below.
- “In addition to long-range interaction and looping functions, characteristic chromatin modifications are found at insulators and are required for insulator activity. Furthermore, RNA molecules are involved in CTCF function. It remains to be seen whether these activities are fundamental to insulator function, or whether they support efficient binding of the architectural proteins, thereby maintaining long-range interactions” (Ali, Renkawitz and Bartkuhn 2016, doi:10.1016/j.gde.2015.11.009).
- “We found transcription to be highly correlated with local chromatin insulation. Therefore, although we confirmed that most TAD [topologically asociated domain] boundaries are conserved, novel borders can occur at promoters of developmentally regulated genes. Furthermore, the correlation between transcription and insulation also extends within TADs. However, we show that activating transcription is not sufficient to cause chromatin insulation, and thus, other factors such as E-P [enhancer-promoter] interactions and specific TFs [transcription factors] likely contribute to creating insulation. Alternatively, changes in chromatin conformation precede and may enable gene expression at specific loci. These findings complement recent results in Drosophila development, which suggested that transcription is not necessary for boundary formation” (Bonev, Cohen, Szabo et al. 2017, doi:10.1016/j.cell.2017.09.043).
-
When bound by various proteins that recognize the particular
insulator sequence, the insulator may serve to:
-
Other DNA regulatory elements
Because of the difficulty of identifying distant-acting regulatory
elements such as activators, silencers, and insulators (see preceding
headings), the known categories of such elements “are likely to be crude
partitions of a vastly more diverse range of regulatory functions” (Noonan
and McCallion 2010).
-
Telomeres
Telomeres, which occur at both ends of chromosomes and consist of
repetitive DNA sequences, tend to be shortened as a result of cell
division and DNA replication, and this shortening is associated with
cellular aging and death. But there are also enzyme molecules that
help to restore the length of shortened telomeres. “It has long been
known that telomeres can silence the expression of nearby genes — a
phenomenon known as the telomere position effect (TPE) — and that
telomere shortening can affect TPE” (Zlotorynski 2014). But now it is
being found that telomeres have a more dynamic and complex role to play
in regulation of gene expression.
- Telomeres facilitate chromosome looping, and this telomere-related looping decreases as telomeres shorten. Researchers showed that in one cell type (myoblasts) genes as far away as 10 megabases from a telomere could have their expression changed by shortening the telomere. Most showed increased expression, but some exhibited the opposite effect. How telomere-chromatin loops are formed and exactly how they bear on the expression of particular genes is not yet known. But it has been shown that there are differences in the high-order organization of chromatin depending on whether a telomere is shorter or longer (Zlotorynski 2014).
- “Robin et al. demonstrate that chromosome looping brings the telomere close to genes up to 10 Mb away from the telomere when telomeres are long, while the loci become separated when telomeres are short. Many loci, including noncoding RNAs, may be regulated by telomere length. This suggests a potential mechanism for how telomere shortening could contribute to aging and disease initiation/progression in human cells long before the induction of a critical DNA damage response” (blurb in Genes and Development for Robin, Ludlow, Batten et al. 2014, doi:10.1101/gad.251041.114).
-
Telomeres
-
DNA methylation
DNA methylation is the addition of methyl groups to DNA bases (most often
cytosine, forming 5-methylcytosine; secondarily adenine). 5-methylcytosine
has been referred to as the “fifth base” of the genome. Millions of bases
are methylated in normal human tissues, with a range of significances that
is hardly less than the significances of the bases themeselves — except
that, unlike the usual case with the four DNA bases, methylation can be
altered during development and in response to environmental influences.
“Dynamic DNA methylation patterns are very important during early
development. During lineage commitment, differentiating cells are thought to
methylate promoters of nontranscribed genes specific to other lineages to
permanently silence them. In contrast, genes that are essential for lineage
specification are kept nonmethylated. DNA methylation–mediated gene
silencing is thought to involve multiple mechanisms that are still not
completely understood. Methylation can directly interfere with the binding
of transcription factors to DNA. Methylated DNA also recruits
transcriptionally repressive methyl-CpG-binding proteins. Furthermore, DNA
methylation can affect nucleosome positioning” (Spruijt and Vermeulen 2014,
doi:10.1038/nsmb.2910).
Referring to DNA methyltransferases (DNMTs):
“It [has been] shown that their catalytic activity is under allosteric
control of N-terminal domains with autoinhibitory function ... Moreover,
targeting and activity of DNMTs were found to be regulated in a concerted
manner by interactors and posttranslational modifications (PTMs) ... We
propose that the allosteric regulation of DNMTs by autoinhibitory domains
acts as a general switch for the modulation of the function of DNMTs,
providing numerous possibilities for interacting proteins, nucleic acids or
PTMs to regulate DNMT activity and targeting. The combined regulation of
DNMT targeting and catalytic activity contributes to the precise
spatiotemporal control of DNMT function and genome methylation in cells”
(Jeltsch and Jurkowska 2016, doi:10.1093/nar/gkw723).
“The classical model of cytosine DNA methylation (the presence of
5-methylcytosine, 5mC) regulation depicts this covalent modification as a
stable repressive regulator of promoter activity. However, whole-genome
analysis of 5mC reveals widespread tissue- and cell type–specific patterns
and pervasive dynamics during mammalian development. Here we review recent
findings that delineate 5mC functions in developmental stages and diverse
genomic compartments as well as discuss the molecular mechanisms that
connect transcriptional regulation and 5mC. Beyond the newly appreciated
dynamics, regulatory roles for 5mC have been suggested in new biological
contexts, such as learning and memory or aging”
(Luo, Hajkova and Ecker 2018, doi:10.1126/science.aat6806).
“Changes in epigenetic DNA methylation are the most promising predictor of
biological age and lifespan in humans, but whether methylation changes
affect ageing is unresolved. Here, we discuss converging data, which
indicate that one mode by which aberrant DNA methylation can affect ageing
is via CCAAT/enhancer binding protein beta (C/EBPβ). This basic
leucine-zipper (bZIP) transcription factor is controlled by the lifespan
regulator mechanistic/mammalian target of rapamycin complex 1 (mTORC1) and
plays an important role in energy homeostasis and adipose tissue
differentiation” (Niehrs and Calkhoven 2019, doi:10.1016/j.tig.2019.11.005).
Complex interaction of multiple factors is illustrated, based on work
in mouse embryonic stem cells:
“The COMPASS protein family catalyzes histone H3 Lys 4 (H3K4) methylation
and its members are essential for regulating gene expression. MLL2/COMPASS
methylates H3K4 on many developmental genes and bivalent clusters ... We
found that MLL2 functions in gene expression by protecting developmental
genes from repression via repelling PRC2 and DNA methylation machineries.
Accordingly, repression in the absence of MLL2 is relieved by inhibition of
PRC2 and DNA methyltransferases. Furthermore, DNA demethylation on such loci
leads to reactivation of MLL2-dependent genes not only by removing DNA
methylation but also by opening up previously CpG methylated regions for
PRC2 recruitment, diluting PRC2 at Polycomb-repressed genes. These findings
reveal how the context and function of these three epigenetic modifiers of
chromatin can orchestrate transcriptional decisions and demonstrate that
prevention of active repression by the context of the enzyme and not H3K4
trimethylation underlies transcriptional regulation on MLL2/COMPASS targets”
(Douillet, Sze, Ryan et al. 2020, doi:10.1038/s41588-020-0618-1).
An investigation of the effect of parasite infection upon the three-spined
stickleback fish: “We showed that the levels of DNA methylation are higher
in infected fish. Results furthermore suggest correlations between DNA
methylation and shifts in key fitness and immune traits between infected and
control fish, including respiratory burst and functional trans-generational
traits such as the concentration of motile sperm. We revealed that genes
associated with metabolic, developmental, and regulatory processes (cell
death and apoptosis) were differentially methylated between infected and
control fish. Interestingly, genes such as the neuropeptide FF receptor 2
and the integrin alpha 1 as well as molecular pathways including the Th1 and
Th2 cell differentiation were hypermethylated in infected fish, suggesting
parasite-mediated repression mechanisms of immune responses. Altogether, we
demonstrate that parasite infection contributes to genome-wide DNA
methylation modifications. Our study ... suggests that epigenetic
mechanisms are complementary to genetic responses against parasite-mediated
selection”
(Sagonas, Meyer, Kaufmann et al. 2020, doi:10.1093/molbev/msaa084).
“The genomes of mammalian neurons are enriched for unique forms of DNA
methylation, including exceptionally high levels of non-CG methylation.
Here, we review recent studies defining how non-CG methylation accumulates
in neurons and is read out by the critical regulator of neuronal
transcription, MeCP2. We discuss the role of gene expression and genome
architecture in establishing non-CG methylation and highlight emerging
mechanistic insights into how non-CG methylation and MeCP2 control
transcription. Further, we describe the cell type-specific functions of this
methylation and explore growing evidence that disruption of this regulatory
pathway contributes to neurodevelopmental disorders. These findings uncover
how the distinctive epigenome in neurons facilitates the development and
function of the complex mammalian brain”
(Clemens and Gabel 2020, doi:10.1016/j.tig.2020.07.009).
- Methylation of gene promoters has long been recognized as repressive of gene expression, although the reality has become steadily more complex. In any case, methylation can repress expression both by directly blocking transcription factors from binding to the promoter, and indirectly via proteins that recognize methylation sites, bind to them, and then prevent RNA polymerase from binding to the promoter.
- In the opposite direction, the binding of transcription factors plays an important role in maintaining methylation (Smith and Meissner 2013).
- CpG methylation reduces DNA backbone flexibility and dynamics (Pennings, Allan and Davey 2005). By changing the mechanical properties of DNA, methylation “is observed to either inhibit or facilitate [DNA] strand separation, depending on methylation level and sequence context” (Severin, Zou, Gaub and Schulten 2011). This has a direct effect on gene expression, since strand separation is essential to the activity of RNA polymerase.
- “Most mammalian RNA polymerase II initiation events occur at CpG islands, which are rich in CpGs and devoid of DNA methylation. Despite their relevance for gene regulation, it is unknown to what extent the CpG dinucleotide itself actually contributes to promoter activity. ... [Our study shows that] CpG density significantly improves motif-based prediction of transcription factor binding. Our experiments also show that high CpG density alone is insufficient for transcriptional activity, yet results in increased transcriptional output when combined with particular transcription factor motifs. However, this CpG contribution to promoter activity is independent of DNA methyltransferase activity” (Li, Gao, Wu et al. 2019, doi:10.1101/gr.240036.118).
- Specialized proteins can bind to methylated DNA and then recruit repressor complexes. Some such proteins may bind to more than one methylated DNA site, resulting in clustering of methylated chromatin.
- Methylation conduces to more regularly spaced nucleosome arrays, and therefore to the formation of densely packed chromatin — all consistent with its generally repressive effects.
- “Nucleosome formation and translational positioning appear to be largely insensitive to DNA (CpG) methylation. There is a small subset of positions, however, that is significantly affected by cytosine methylation”. Such methylation may conduce to more regularly spaced nucleosome arrays and therefore to the formation of densely packed chromatin (Pennings 2005). Also, “nucleosomes assembled with non-methylated DNA appear less stable than those assembled with mDNA [methylated DNA]” (Severin, Zou, Gaub and Schulten 2011).
- DNA methylation patterns vary from regulatory regions to gene promoters to gene bodies to repetitive elements, “suggesting that different mechanisms could be involved in the regulation of [methylation] across the genome and in the interaction with chromatin-associated proteins and histone modifications” (Bell and Spector 2011).
- The end result of DNA methylation can in some cases be enhanced rather than suppressed gene expression. For example, an imprinting control element that is a long-range cis-acting repressor of its associated genes can itself be repressed by DNA methylation. In this way methylation activates expression of imprinted genes that are repressed by default.
- Generally, DNA methylation within a gene, and especially in its promoter region, has been thought to suppress expression of the gene. More recently, methylation of the first exon has been found to be more directly correlated with silencing gene transcription than is promoter methylation (Brenet, Moh, Funk et al. 2011).
- “Consistent with previous work we found that intragenic methylation is positively correlated with gene expression and that exons are more highly methylated than their neighboring intronic environment. Intriguingly, in this study we identified a unique subset of hypomethylated exons that demonstrate significantly lower methylation levels than their surrounding introns. Furthermore, we observed a negative correlation between exon methylation and the density of the majority of histone modifications. Specifically, we demonstrate that hypo-methylated exons at highly expressed genes are associated with open chromatin and have a characteristic histone code comprised of significantly high levels of histone markings. Overall, our comprehensive analysis of the human exome supports the presence of regulatory hypomethylated exons in protein coding genes. In particular our results reveal a previously unrecognized diverse and complex role of the epigenetic landscape within the gene body” (Singer, Kosti, Pachter and Mandel-Gutfreund 2015, doi:10.1093/nar/gkv153).
- “Gene-body methylation inhibits transcription initiation from cryptic promoters” (Spruijt and Vermeulen 2014, doi:10.1038/nsmb.2910).
- “Our results ... confirm a previous finding that methylation signals at transcript bodies are more indicative of gene expression levels than promoter methylation signals” (Li, Gao, Wu et al. 2019, doi:10.1101/gr.240036.118).
- There appears to be “a major role for intragenic methylation in regulating cell context-specific alternative promoters in gene bodies” (Maunakea, Nagarajan, Bilenky et al. 2010).
- DNA methylation can also suppress the expression of microRNAs, which in turn play a vital role in gene regulation.
- DNMT1-directed RNA: Expression of the human CEPBA gene correlates with the separate expression of a long noncoding RNA (ecCEBPA) that encompasses the entire mRNA sequence and more. Apparently, the long noncoding RNA anchors itself to the gene locus and also becomes attched to DNA methyltransferase 1 (DNMT1), an enzyme that methylates DNA. In this way, methylation of the gene is prevented, and the gene can be expressed. It is proposed that DNMT1 recognizes and binds to the long noncoding RNA by virtue of the latter’s secondary structure (a stem loop), and that the noncoding RNA binds to the gene locus via a “locus-selective triplex/quadruplex”. (See RNA structure and dynamics under Other Aspects Of the Molecular Structure and Dynamics Of DNA and RNA below.) The authors of this study (Di Ruscio, Ebralidze, Benoukraf et al. 2013) went on to identify a “large set” of genes subject to regulation by means of noncoding DNA–RNA–DNMT1 binding.
- DNA methylation is tissue-specific. For example, non-CpG DNA methylation is much more widespread in pluripotent cells than in somatic cells generally (and is also highly variable in those pluripotent cells). And a study of neural precursor cells showed that hypomethylated regions (with CpG methylation levels of 10–50%) are specific to particular cell types. This specificity was found in at least some cases to correlate with protein transcription factors and regulators bound to the hypomethylated regions. (See original research cited in Stower 2012).
- “Interactions with methylated DNA are highly dynamic during cellular differentiation” (Spruijt and Vermeulen 2014, doi:10.1038/nsmb.2910).
- There is intimate interaction (mutual regulation) between DNA methylation and histone modifications.
- Small interfering RNAs contribute to methylation.
- “Although DNA methylation was originally thought to only affect transcription, emerging evidence shows that it also regulates alternative splicing. Exons, and especially splice sites, have higher levels of DNA methylation than flanking introns, and the splicing of about 22% of alternative exons is regulated by DNA methylation. Two different mechanisms convey DNA methylation information into the regulation of alternative splicing. The first involves modulation of the elongation rate of RNA polymerase II (Pol II) by CCCTC-binding factor and methyl-CpG binding protein 2; the second involves the formation of a protein bridge by heterochromatin protein 1 (HP1) that recruits splicing factors onto transcribed alternative exons. These two mechanisms, however, regulate only a fraction of such events, implying that more underlying mechanisms remain to be found” (Maor, Yearim and Ast 2015, doi:10.1016/j.tig.2015.03.002).
- “In conjunction with accumulation of genetic lesions, there is an aberrant pattern for the different epigenetic effectors: DNA methylation, histone modifications, and miRNAs. In normal cells, the interplay between the epigenetic factors and the chromatin structure leads to a tuned gene regulation. However, in cancer cells tumor suppressor gene promoters become hypermethylated and with an altered global pattern of histone modifications resulting in aberrant gene silencing. Moreover, global hypomethylation leads to chromosome instability and fragility. Epigenetic changes, including DNA methylation and histone modifications are responsible for abnormal mRNA and miRNA expression producing altered activation of oncogenes and silencing of tumor suppressor genes” (doi:10.1016/j.gde.2012.02.008).
- Dynamically regulated DNA methylation: In a review article, Jeltsch and Jurkowska (2014) point to inadequacies in the prevailing model of DNA methylation, which, they say, leaves out “substantial experimental evidence from the past decade” that suggests the need for a more dynamic view. According to this view, “DNA methylation at each site is determined by the local activity of DNA methyltransferases (Dnmts), DNA demethylases, and the DNA replication rate”. Further, “DNA methylation is guided by an epigenetic network, in which DNA modifications, histone tail modifications, and other epigenetic marks influence each other and function in a synergistic fashion. These marks recruit Dnmts [DNA methyltransferases] and DNA demethylases to DNA regions, which are targets of methylation and demethylation, simultaneously reducing their binding to other parts, and regulate the activity of the enzymes. ... The average DNA methylation level of DNA regions is inherited rather than the methylation state of individual CpG sites”. Other considerations lead the authors to say that “the overall process of DNA methylation is more complicated than anticipated by the classical maintenance model”.
- In the same vein: “We conducted a comprehensive survey involving multiple cell lines, TFs, and methylation types and found that there are intimate relationships between TF binding and methylation level changes around the binding sites” (Xu, Li, Zhao et al. 2015, doi:10.1093/nar/gkv151).
- There are many trait differences between men and women, and now a study raises the question, How many of these differences are related to differential DNA methylation? “We identified 1184 CpGs showing stable DNA methylation differences between men and women in four European cohorts. These sites were found to be enriched at CpG island shores and at imprinted genes. Furthermore, we observed enrichment for three gene ontology terms. [“Gene ontology” refers to a database of standardized descriptors of gene attributes and functions.] From these results, we conclude that sex-dependent DNA methylation may be implicated in the observed sex discordance in various traits and diseases. Functional associations were demonstrated through mRNA expression analysis, which revealed two genes with significant sex- and DNA methylation-dependent expression differences” (Singmann, Shem-Tov, Simone Wahl et al. 2015, doi:10.1186/s13072-015-0035-3).
- “We provide evidence that estrogen receptor beta (ERβ) plays a role in regulating DNA methylation at specific genomic loci, likely as the result of its interaction with TDG [thymine DNA glycosylase] at these regions. Our findings imply a novel function of ERβ, beyond direct transcriptional control, in regulating DNA methylation at target genes. Further, they shed light on the question how DNA methylation is regulated at specific genomic loci by supporting a concept in which sequence-specific transcription factors can target factors that regulate DNA methylation patterns” (Liu, Duong, Krawczyk et al. 2016, doi:10.1186/s13072-016-0055-7).
- “In vertebrates, methylation of cytosine at CpG sequences is implicated in stable and heritable patterns of gene expression. The classical model for inheritance, in which individual CpG sites are independent, provides no explanation for the observed non-random patterns of methylation ... we show a strong dependence of methylation on the number and density of CpG organization. CpG clusters with fewer, or less densely spaced, CpGs are predominantly hyper-methylated, while larger clusters are predominantly hypo-methylated. Intermediate clusters, however, are either hyper- or hypo-methylated but are rarely found in intermediate methylation states. We develop a model for spatially-dependent collaboration between CpGs, where methylated CpGs recruit methylation enzymes that can act on CpGs over an extended local region, while unmethylated CpGs recruit demethylation enzymes that act more strongly on nearby CpGs. This model can reproduce the effects of CpG clustering on methylation and produces stable and heritable alternative methylation states of CpG clusters, thus providing a coherent model for methylation inheritance and methylation patterning” (Lövkvist, Dodd, Sneppen and Haerter 2016, doi:10.1093/nar/gkw124).
- “Remodeling DNA methylation in mammalian genomes can be global, as seen in preimplantation embryos and primordial germ cells (PGCs), or locus specific, which can regulate neighboring gene expression. In PGCs, global and locus-specific DNA demethylation occur in sequential stages, with an initial global decrease in methylated cytosines (stage I) followed by a Tet methylcytosine dioxygenase (Tet)-dependent decrease in methylated cytosines that act at imprinting control regions and meiotic genes (stage II) ... Here we show that Dnmt1 [the enzyme DNA (cytosine-5)-methyltransferase 1] preserves DNA methylation through stage I at imprinting control regions and meiotic gene promoters and is required for the pericentromeric enrichment of 5hmC [5-hydroxymethylcytosine]. We discovered that the functional consequence of abrogating two-stage DNA demethylation in PGCs was precocious germline differentiation leading to hypogonadism and infertility. Therefore, bypassing stage-specific DNA demethylation has significant consequences for progenitor germ cell differentiation and the ability to transmit DNA from parent to offspring. (Hargan-Calvopina, Taylor, Cook et al. 2016, doi:10.1016/j.devcel.2016.07.019).
- “We show that, in mouse embryonic stem cells, Dnmt3b-dependent intragenic DNA methylation protects the gene body from spurious RNA polymerase II entry and cryptic transcription initiation. Using different genome-wide approaches, we demonstrate that this Dnmt3b function is dependent on its enzymatic activity and recruitment to the gene body by H3K36me3. Furthermore, the spurious transcripts can either be degraded by the RNA exosome complex or capped, polyadenylated, and delivered to the ribosome to produce aberrant proteins. Elongating RNA polymerase II therefore triggers an epigenetic crosstalk mechanism that involves SetD2, H3K36me3, Dnmt3b and DNA methylation to ensure the fidelity of gene transcription initiation, with implications for intragenic hypomethylation in cancer” (Neri, Rapelli, Krepelova et al. 2017, doi:10.1038/nature21373).
- “Methylation in gene bodies prevents aberrant and potentially deleterious intragenic transcription” (Zlotorynski 2017, doi:10.1038/nrm.2017.25).
- “DNA methylation is a key regulator of embryonic stem cell (ESC) biology, dynamically changing between naïve, primed, and differentiated states. The p53 tumor suppressor is a pivotal guardian of genomic stability, but its contributions to epigenetic regulation and stem cell biology are less explored. We report that, in naïve mouse ESCs (mESCs), p53 restricts the expression of the de novo DNA methyltransferases Dnmt3a and Dnmt3b while up-regulating Tet1 and Tet2, which promote DNA demethylation. The DNA methylation imbalance in p53-deficient (p53–/–) mESCs is the result of augmented overall DNA methylation as well as increased methylation landscape heterogeneity. In differentiating p53–/– mESCs, elevated methylation persists, albeit more mildly. Importantly, concomitant with DNA methylation heterogeneity, p53–/– mESCs display increased cellular heterogeneity both in the ‘naïve’ state and upon induced differentiation. This impact of p53 loss on 5-methylcytosine (5mC) heterogeneity was also evident in human ESCs and mouse embryos in vivo. Hence, p53 helps maintain DNA methylation homeostasis and clonal homogeneity, a function that may contribute to its tumor suppressor activity” (Tovy, Spiro, McCarthy et al. 2017, doi:10.1101/gad.299198.117).
- “We report locus-specific disintegration of megabase-scale chromosomal conformations in brain after neuronal ablation of Setdb1 (also known as Kmt1e; encodes a histone H3 lysine 9 methyltransferase), including a large topologically associated 1.2-Mb domain conserved in humans and mice that encompasses >70 genes at the clustered protocadherin locus (hereafter referred to as cPcdh). The cPcdh topologically associated domain (TADcPcdh) in neurons from mutant mice showed abnormal accumulation of the transcriptional regulator and three-dimensional genome organizer CTCF at cryptic binding sites, in conjunction with DNA cytosine hypomethylation, histone hyperacetylation and upregulated expression. Genes encoding stochastically expressed protocadherins were transcribed by increased numbers of cortical neurons, indicating relaxation of single-cell constraint. SETDB1-dependent loop formations bypassed 0.2–1 Mb of linear genome and radiated from the TADcPcdh fringes toward cis-regulatory sequences within the cPcdh locus, counterbalanced shorter-range facilitative promoter–enhancer contacts and carried loop-bound polymorphisms that were associated with genetic risk for schizophrenia. We show that the SETDB1 repressor complex, which involves multiple KRAB zinc finger proteins, shields neuronal genomes from excess CTCF binding and is critically required for structural maintenance of TADcPcdh” (Jiang, Loh, Rajarajan 2017, doi:10.1038/ng.3906).
- “Mutual antagonism between DNA methylation and H3K27me3 histone methylation suggests a dynamic crosstalk between these epigenetic marks that could help ensure correct gene expression programmes. Work from Manzo et al (2017) now shows that an isoform of de novo DNA methyltransferase DNMT3A provides specificity in the system by depositing DNA methylation at adjacent “shores” of hypomethylated bivalent CpG islands (CGI) in mouse embryonic stem cells (mESCs). DNMT3A1‐directed methylation appears to be instructive in maintaining the H3K27me3 profile at the hypomethylated bivalent CGI promoters of developmentally important genes”. (Meehan and Pennings 2017, doi:10.15252/embj.201798498)
- “Cytosine DNA methylation is a heritable and essential epigenetic mark. During DNA replication, cytosines on mother strands remain methylated, but those on daughter strands are initially unmethylated. These hemimethylated sites are rapidly methylated to maintain faithful methylation patterns. Xu and Corces mapped genome-wide strand-specific DNA methylation sites on nascent chromatin, confirming such maintenance in the vast majority of the DNA methylome. However, they also identified a small fraction of sites that were stably hemimethylated and showed their inheritance at CTCF (CCCTC-binding factor)/cohesin binding sites. These inherited hemimethylation sites were required for CTCF and cohesin to establish proper chromatin interactions” (Xu and Corces 2018, doi:10.1126/science.aan5480). “This challenges the prevailing view that hemimethylation is transient and suggests that this DNA modification could be maintained as a stable epigenetic state” (Table of Contents blurb in Science, March 9, 2018).
- “Our integrative analysis clearly reveals the important and conserved role of the methylation level of the first intron and its inverse association with gene expression regardless of tissue and species”. “Notably, the first intron exhibits a tissue-independent enrichment for TF-binding motifs and the methylation of the CpGs they contain is indicative of the gene expression level. Furthermore, the first intron presents a higher number of tDMRs [tissue-specific differentially methylated regions] than other gene features, suggestive of a regulatory role in tissue-specific expression” (Anastasiadi, Esteve-Codina and Piferrer 2018, doi:10.1186/s13072-018-0205-1).
- “Histone methylation is required for the establishment and maintenance of gene expression patterns that determine cellular identity, and its perturbation often leads to aberrant development and disease. Recruitment of histone methyltransferases (HMTs) to gene regulatory elements (GREs) of developmental genes is important for the correct activation and silencing of these genes, but the drivers of this recruitment are largely unknown. Here we propose that lineage-instructive transcription factors (Lin-TFs) act as general recruiters of HMT complexes to cell type-specific GREs through protein–protein interactions. We also postulate that the specificity of these interactions is dictated by Lin-TF post-translational modifications (PTMs), which act as a ‘transcription factor code’ that can determine the directionality of cell fate decisions during differentiation and development” (Garcia and Graf 2021, doi:10.1016/j.tcb.2021.04.001).
- “Epigenetic modifications on the chromatin do not occur in isolation. Chromatin-associated proteins and their modification products form a highly interconnected network, and disturbing one component may rearrange the entire system ... It is important to understand the rules governing epigenetic interactions. Here, we use the mouse embryonic stem cell (mESC) model to describe in detail the relationships within the H3K27-H3K36-DNA methylation subnetwork. In particular, we focus on the major epigenetic reorganization caused by deletion of the histone 3 lysine 36 methyltransferase NSD1, which in mESCs deposits nearly all of the intergenic H3K36me2. Although disturbing the H3K27 and DNA methylation (DNAme) components also affects this network to a certain extent, the removal of H3K36me2 has the most drastic effect on the epigenetic landscape, resulting in full intergenic spread of H3K27me3 and a substantial decrease in DNAme. By profiling DNMT3A and CHH methylation (mCHH), we show that H3K36me2 loss upon Nsd1-KO [Nsd1 knockout] leads to a massive redistribution of DNMT3A and mCHH away from intergenic regions and toward active gene bodies, suggesting that DNAme reduction is at least in part caused by redistribution of de novo methylation. Additionally, we show that pervasive acetylation of H3K27 is regulated by the interplay of H3K36 and H3K27 methylation” (Chen, Hu, Horth et al. 2022, doi:10.1101/gr.276383.121)
-
DNA 5-hydroxymethylation
Recent work is rapidly opening a window onto another kind of DNA
modification. Methylated (5-methylcytosine) sites on DNA can be converted
to 5-hydroxymethylated sites (5-hydroxymethylcytosine, or 5hmC), and 5hmC
has been referred to as the “sixth base” of the genome (where
5-methylcytosine is regarded as the “fifth base"; see under “DNA
methylation” above).
“Oxidation of 5mC appears to be a step in several active DNA demethylation
pathways, which may be important for normal processes, as well as global
hypomethylation during cancer development and progression” (Kinney, Pradhan
2013).
However, an article entitled “De novo DNA methylation drives 5hmC
accumulation in mouse zygotes” in Nature Cell Biology has this
description attached: “Hajkova and colleagues show that 5mC loss and 5hmC
accumulation are uncoupled during zygotic epigenetic reprogramming”
(Amouroux, Nashun, Shirane et al. 2016, doi:10.1038/ncb3296).
“Epigenetic factors underlie cellular identity through the regulation of
transcriptional networks that establish a cell’s phenotype and function.
Cell conversions are directed by transcription factor binding at target DNA
which induce changes to identity-specific gene regulatory programs. The
degree of cell plasticity is determined by the interplay of epigenetic
mechanisms to create a landscape susceptible to such binding events.
5-hydroxymethylcytosine, a key intermediate during the process of DNA
demethylation, is an epigenetic modification involved in controlling these
epigenetic dynamics related to cell identity”
(Honig and Murrell 2025, doi:10.1186/s13072-025-00601-w).
- There is “evidence for a role of 5hmC in both transcriptional activation and repression,” and this occurs “in a context-dependent manner”. 5hmC appears to perform its role at least in part by “establishing and maintaining chromatin structure” (Wu, D’Alessio, Ito et al. 2011).
- “We find that global 5hmC content of normal human tissues is highly variable, does not correlate with global 5-methylcytosine content, and decreases rapidly as cells from normal tissue adapt to cell culture...we find 5hmC associated primarily, but not exclusively, with the body of transcribed genes, and that within these genes 5hmC levels are positively correlated with transcription levels”. But this correlation is greatly outweighed by the tissue-type correlation: “a gene transcribed at a similar level in several different tissues may have vastly different levels of 5hmC (>20-fold) dependent on tissue type”. All this suggests that “the functional importance of 5hmC varies between tissues” (Nestor, Ottaviano, Reddington et al. 2012).
- “We show by genome-wide mapping that the newly discovered deoxyribonucleic acid (DNA) modification 5-hydroxymethylcytosine (5hmC) is dynamically associated with transcription factor binding to distal regulatory sites during neural differentiation of mouse P19 cells and during adipocyte differentiation of mouse 3T3-L1 cells...distal regions gaining 5hmC together with H3K4me2 and H3K27ac in P19 cells behave as differentiation-dependent transcriptional enhancers. Identified regions are enriched in motifs for transcription factors regulating specific cell fates...kinetic studies of cytosine hydroxymethylation of selected enhancers indicated that DNA hydroxymethylation is an early event of enhancer activation. Hence, acquisition of 5hmC in cell-specific distal regulatory regions may represent a major event of enhancer progression toward an active state and participate in selective activation of tissue-specific genes” (Sérandour, Avner, Oger et al. 2012).
- “Genome-wide mapping reveals a demolished 5-hydroxymethylcytosine landscape in human melanoma epigenome” and “Loss of 5-hmC is an epigenetic hallmark of melanoma, with diagnostic/prognostic value” (Lian, Xu, Ceol et al. 2012).
- Hydroxylation of DNA methylation to 5hmC (via the hydroxylases, TET1 and TET2) has been shown to facilitate the establishment of (induced) pluripotency, helping to “overcome epigenetic roadblocks during reprogramming and transdifferentiation” (Costa, Ding, Theunissen et al. 2013). However, still more recent research begins to show a more complex situation: “TET1 either positively or negatively regulates somatic cell reprogramming depending on the absence or presence of vitamin C. ... Our findings suggest that vitamin C has a vital role in determining the biological outcome of TET1 function at the cellular level” (Chen, Guo, Zhang et al. 2013a).
- Study on mammalian embryonic stem cells: “We demonstrate that [the deacetylase] SIRT6 functions as a chromatin regulator safeguarding the balance between pluripotency and differentiation through Tet-mediated production of 5-hydroxymethylation” (Etchegaray, Chavez, Yun Huang et al. 2015, doi:10.1038/ncb3147).
- “Tet1-mediated DNA hydroxymethylation plays a critical role in the epigenetic regulation of the Wnt pathway in intestinal stem and progenitor cells and consequently in the self-renewal of the intestinal epithelium” (Kim, Sheaffer, Choi et al. 2016, doi:10.1101/gad.288035.116).
- This item doesn’t really belong here, since it involves histone acetylation, not DNA hydroxymethylation. But it usefully illustrates a general principle of the organism: nothing has just one function. “Recent studies have demonstrated that Tet1 could modulate transcriptional expression independent of its DNA demethylation activity ... Here, we uncovered that Tet1 formed a chromatin complex with histone acetyltransferase Mof and scaffold protein Sin3a in mouse embryonic stem cells ... Tet1 facilitated chromatin affinity and enzymatic activity of hMOF against acetylation of histone H4 at lysine 16 via preventing auto-acetylation of hMOF, to regulate expression of the downstream genes, including DNA repair genes. We found that Tet1 knockout MEF [mouse embryonic fibroblast] cells exhibited an accumulation of DNA damage and genomic instability and Tet1 deficient mice were more sensitive to x-ray exposure. Taken together, our findings reveal that Tet1 forms a complex with hMOF to modulate its function and the level of H4K16Ac ultimately affect gene expression and DNA repair” (Zhong, Li, Cai et al. 2017, doi:10.1093/nar/gkw919).
- “We show ... that TET2 localizes to regions of open chromatin and cell-type–specific enhancers. We find that deletion of Tet2 in native hematopoiesis as well as fully transformed acute myeloid leukemia results in changes in transcription factor (TF) activity within these regions, and we provide evidence that loss of TET2 leads to attenuation of chromatin binding of members of the basic helix–loop–helix TF family. Together, these findings demonstrate that TET2 activity shapes the local chromatin environment at enhancers to facilitate TF binding and provides an example of how epigenetic dysregulation can affect gene expression patterns and drive disease development” (Rasmussen, Berest, Keβler et al. 2019, doi:10.1101/gr.239277.118).
- “A new study demonstrates that 5-hydroxymethylcytosine serves to counteract inappropriate, spurious intragenic transcription in airway smooth muscle cells and by doing so, this DNA base functions in the prevention of chronic inflammation in the lung and an asthma-like phenotype” (Pfeifer 2023, doi:10.1038/s41588-022-01253-2).
-
DNA N6-methyladenine
“The recent discovery of N6-methyladenine (N6-mA) in
mammalian genomes suggests that it may serve as an epigenetic regulatory
mechanism ... Here we show that N6-mA has a key role in changing
the epigenetic landscape during cell fate transitions in early development.
We found that N6-mA is upregulated during the development of
mouse trophoblast stem cells, specifically at regions of stress-induced DNA
double helix destabilization (SIDD). Regions of SIDD are conducive to
topological stress-induced unpairing of the double helix and have critical
roles in organizing large-scale chromatin structures. We show that the
presence of N6-mA reduces the in vitro interactions by more than
500-fold between SIDD and SATB1, a crucial chromatin organizer that
interacts with SIDD regions. Deposition of N6-mA also antagonizes
SATB1 function in vivo by preventing its binding to chromatin. Concordantly,
N6-mA functions at the boundaries between euchromatin and
heterochromatin to restrict the spread of euchromatin. Repression of
SIDD–SATB1 interactions mediated by N6-mA is essential for gene
regulation during trophoblast development in cell culture models and in
vivo. Overall, our findings demonstrate an unexpected molecular mechanism
for N6-mA function via SATB1, and reveal connections between DNA
modification, DNA secondary structures and large chromatin domains in early
embryonic development”
(Li, Zhao, Nelakanti et al. 2020, doi:10.1038/s41586-020-2500-9).
-
Nucleosome positioning
First, note the larger context, of which nucleosome positioning is only one
aspect: “Nucleosome dynamics are governed by a complex interplay of histone
composition, histone post-translational modifications, nucleosome occupancy
and positioning within chromatin, which are influenced by numerous
regulatory factors, including general regulatory factors, chromatin
remodellers, chaperones and polymerases. It is now known that these dynamics
regulate diverse cellular processes ranging from gene transcription to DNA
replication and repair”
(Lai and Pugh 2017, doi:10.1038/nrm.2017.47).
Nucleosomes are DNA-enwrapped histone protein complexes. The core
histones can slide along the DNA (or the DNA can be pulled around the
histones). The DNA typically makes about 1.67 turns around the histone
core. There are some 30 million nucleosomes helping to give structure to
the human genome, and 75-90% of eukaryotic genomic DNA is said to be
wrapped around nucleosomes.
“The genome-wide pattern of nucleosome positioning is determined by the
combination of DNA sequence, ATP-dependent nucleosome remodeling enzymes
and transcription factors that include activators, components of the
preinitiation complex and elongating RNA polymerase II. These determinants
influence each other such that the resulting nucleosome positioning
patterns are likely to differ among genes and among cells in a population,
with consequent effects on gene expression” (Struhl and Segal 2013).
“Nucleosomes around transcriptional start sites in silent parts of the
genome are regularly spaced, but their position between individual cells
varies considerably. In contrast, nucleosomes at start sites in actively
transcribed regions have more heterogeneity in spacing but are positioned
more precisely. These would appear to represent differences in chromatin
stability and the activity of chromatin remodeling factors in the active
regions. The authors show that spacing between nucleosomes is bimodal, with
peaks at 190bp and 300 bp, which correlate with differential accessibility.
Moreover, variation in gene expression is shown to be correlated to
variation in nucleosome positioning”
(Kruger 2018, doi:10.1016/j.cell.2018.11.001).
“Chromatin remodellers [that is, ATP-dependent molecular complexes that
establish and maintain the positions of nucleosomes] can be stratified into
two functional groups. Group 1 (BRG1, SNF2H, CHD3 and CHD4) has a clear
preference for binding at ‘actively marked’ chromatin and Group 2 (BRM,
INO80, SNF2L and CHD1) for ‘repressively marked’ chromatin. We find that
histone modifications and chromatin architectural features, but not DNA
methylation, stratify the remodellers into these functional groups”
(Giles, Gould, Du et al. 2019, doi:10.1186/s13072-019-0258-9).
“Nucleosome turnover rates are modulated by a variety of factors, including
DNA sequence, histone variant composition, histone chaperone availability,
the local chromatin context (comprising histone density and ATP-dependent
chromatin remodeller activity) and occupancy by linker histones and TFs.
Nucleosome preference for particular DNA sequences is well established in
vitro and in yeast; however, this bias is less pronounced in multicellular
systems, and it is weakly predictive of human nucleosome positioning”
(Klemm, Shipony and Greenleaf 2019, doi:10.1038/s415760180089-8).
(In yeast:) “We show that many promoters are affected by multiple CRs
[chromatin remodeler enzymes] that operate in concert or in opposition to
position the key transcription start site-associated +1 nucleosome. We
also show that nucleosome movement after CR inactivation usually results
from the activity of another CR and that in the absence of any remodeling
activity, +1 nucleosomes largely maintain their positions. Finally, we
present functional assays suggesting that +1 nucleosome positioning often
reflects a trade-off between maximizing RNA polymerase recruitment and
minimizing transcription initiation at incorrect sites” (Kubik, Bruzzone,
Challal et al. 2019, doi:10.1038/s41594-019-0273-3).
“Mechanical deformations of DNA such as bending are ubiquitous and have been
implicated in diverse cellular functions. [In Saccharomyces
cerevisiae] we found sequence-encoded regions of unusually low
bendability within nucleosome-depleted regions upstream of transcription
start sites (TSSs). Low bendability of linker DNA inhibits nucleosome
sliding into the linker by the chromatin remodeller INO80, which explains
how INO80 can define nucleosome-depleted regions in the absence of other
factors. Chromosome-wide, nucleosomes were characterized by high DNA
bendability near dyads and low bendability near linkers. This contrast
increases for deeper gene-body nucleosomes but disappears after random
substitution of synonymous codons, which suggests that the evolution of
codon choice has been influenced by DNA mechanics around gene-body
nucleosomes. Furthermore, we show that local DNA mechanics affect
transcription through TSS-proximal nucleosomes. Overall, this genome-scale
map of DNA mechanics indicates a ‘mechanical code’ with broad functional
implications”
(Basu, Bobrovnikov, Qureshi et al. 2021, doi:10.1038/s41586-020-03052-3).
- Removal of nucleosomes from promoters and the positioning of nucleosomes downstream from promoters (in gene bodies) “play crucial roles in determining the transcription level, cell-to-cell variation, activation and repression dynamics, and might also function in defining the start and end points of transcribed regions. Nucleosomes affect transcription mostly by modulating the accessibility of regulatory factors [which come in vast variety] and the transcriptional machinery of the underlying DNA sequence” (Bai and Morozov 2010).
- It’s been known that nucleosome positioning bears on transcription initiation and termination. Now, in a study on yeast, it’s been shown to bear on transcription elongation as well. There is “a strong dependency of RNA pol[ymerase] II elongation activity on nucleosome positioning. Such nucleosome dependence causes gene-specific profiles and reveals that RNA pol II-dependent genes differ not only at the transcription initiation level, as generally acknowledged, but also at the elongation level. This novel perspective involves inactivation/reactivation as an important aspect of RNA polymerase dynamics throughout the transcription cycle” (Jordán-Pla, Gupta, de Miguel-Jiménez et al. 2015, doi:10.1093/nar/gku1349).
- The DNA double helix is a rather stiff molecule, and its local structure — for example, its bendability or flexibility — apparently plays a substantial role in determining how and where it can wrap around the nucleosome core particle (histone octomer). Also, according to a study in fruit flies and other organisms, some “preferential positions for nucleosomes were found where the mean helical rise [the length of the double helix per given number of base pairs] reaches its largest values at GC-rich DNA sequences” (Pedone and Santoni 2012). That is, positioning of certain nucleosomes is favored where the DNA is most “stretched”.
- Nucleosomes play an important role in chromatin packaging, which in turn is important for gene expression.
- As little as a two or three base-pair shift in the position of a nucleosome over a gene promoter can make the difference between expression or silence of the gene (Martinez-Campa 2004).
- Removal of a nucleosome via disassembly of its histone core particle not only removes an obstacle to DNA access, but also facilitates untwisting of the DNA (because the single negative supercoil constrained by the core particle is released) and therefore facilitates transcription initiation.
- Nucleosome positioning tends to protect DNA from DNA methylation, and therefore presumably engages in crosstalk with all the functions of DNA methylation (Felle, Hoffmeister, Rothammer et al. 2011). See DNA methylation above.
-
Using the highest-resolution imaging techniques employed to date with
living cells, one group of researchers reports: “Our observations
indicate that nucleosomes are grouped in discrete domains along the
chromatin fiber, which we termed ‘nucleosome clutches’ ... Clutches are
interspersed with nucleosome-depleted regions and the number of
nucleosomes per clutch is very heterogeneous in a given nucleus arguing
against the existence of a well-organized and ordered fiber ... [The
techniques employed] showed increased levels of H1 [linker histones] in
larger and denser clutches containing more nucleosomes, which formed the
‘closed’ heterochromatin. On the other hand, ‘open’ chromatin was formed
by smaller and less dense clutches which associated with RNA Polymerase
II. Strikingly, despite the heterogeneity in clutch size in a given
nucleus, on average differentiated cells contained larger and denser
clutches compared to stem cells” (Ricci, Manzo, García-Parajo et al.
2015, doi:10.1016/j.cell.2015.01.054).
Bear in mind that there is actually no evidence that the chromatin fiber is not “well-organized”; everything suggests it is wonderfully fine-tuned for the infinitely nuanced expression of thousands of genes. It’s just that it isn’t “military order”; rather, it’s more like an intricately choreographed dance.
- Example of a nucleosome remodeling factor’s regulation of nucleosome positioning: The esBAF (SWI/SNF-family) remodeling factor “suppresses transcription of noncoding RNAs from ∼57,000 nucleosome-depleted regions (NDRs) throughout the genome of mouse embryonic stem cells”. esBAF functions both to (1) keep NDRs nucleosome-free, and (2) promote elevated nucleosome occupancy adjacent to NDRs, regardless of the occupancy within the NDRs. But it turns out that only (2) is required for suppressing transcription of noncoding RNAs. This suggests that the flanking nucleosomes form a barrier to pervasive transcription. “This mechanism is fundamentally distinct from the well-established role of esBAF as an activator of gene expression, which is thought to function by increasing chromatin accessibility” (Hainer, Gu, Carone et al. 2015, doi:10.1101/gad.253534.114).
- “In zebrafish, DNA methylation patterns are programmed in transcriptionally quiescent cleavage embryos; paternally inherited patterns are maintained, whereas maternal patterns are reprogrammed to match the paternal. Here, we provide the mechanism by demonstrating that “Placeholder” nucleosomes, containing histone H2A variant H2A.Z(FV) and H3K4me1, virtually occupy all regions lacking DNA methylation in both sperm and cleavage embryos and reside at promoters encoding housekeeping and early embryonic transcription factors. Upon genome-wide transcriptional onset, genes with Placeholder become either active (H3K4me3) or silent (H3K4me3/K27me3). Notably, perturbations causing Placeholder loss confer DNA methylation accumulation, whereas acquisition/expansion of Placeholder confers DNA hypomethylation and improper gene activation. Thus, during transcriptionally quiescent gametic and embryonic stages, an H2A.Z(FV)/H3K4me1-containing Placeholder nucleosome deters DNA methylation, poising parental genes for either gene-specific activation or facultative repression” (Murphy, Wu, James et al. 2018, doi:10.1016/j.cell.2018.01.022).
- “Here, we conducted a genome-wide comparison of high-resolution nucleosome maps in peripheral blood B cells from patients with chronic lymphocytic leukemia (CLL) and healthy individuals at single-base-pair resolution. Our investigation uncovered significant changes of nucleosome positioning in CLL. Globally, the spacing between nucleosomes—the nucleosome repeat length (NRL)—is shortened in CLL. This effect is stronger in the more aggressive IGHV-unmutated CLL subtype than in the IGHV-mutated CLL subtype. Changes in nucleosome occupancy at specific sites are linked to active chromatin remodeling and reduced DNA methylation. Nucleosomes lost or gained in CLL marks differential binding of 3D chromatin organizers such as CTCF as well as immune response–related transcription factors and delineated mechanisms of epigenetic deregulation. The principal component analysis of nucleosome occupancy in cancer-specific regions allowed the classification of samples between cancer subtypes and normal controls. Furthermore, patients could be better assigned to CLL subtypes according to differential nucleosome occupancy than based on DNA methylation or gene expression. Thus, nucleosome positioning constitutes a novel readout to dissect molecular mechanisms of disease progression and to stratify patients” (Piroeva, McDonald, Xanthopoulos et al. 2023, doi:10.1101/gr.277298.122).
-
Histone displacement and replacement during elongation
As the body of a gene is being transcribed (a process called
“elongation”), the nucleosomes along this stretch of DNA are an impediment
to the transcribing enzyme. The histones constituting the core of the
nucleosome need to be displaced so that transcription can proceed along the
whole length of the gene. (However, there is still a good deal of
unclarity about exactly what happens with nucleosomes during
transcription.)
Processes for modulating the nucleosome barrier “fall into three broad
classes: mechanisms that alter nucleosomes (chromatin modifiers),
mechanisms that mobilize nucleosomes (chromatin remodelers), and mechanisms
that facilitate Pol II [RNA polymerase II] activity (elongation factors).
Recently, the structure of DNA itself has emerged as a mediator of
nucleosome dynamics that can also affect the strength of the barrier”
(Teves, Weber and Henikoff 2014).
“Many questions still remain to be
answered. For instance, the discovery that the nucleosome barrier in
vivo is context-specific, with the +1 nucleosome posing the strongest
barrier, raises several questions. What determines the context-specificity
of the +1 nucleosome? Are the mechanisms for overcoming the +1 nucleosomal
barrier distinct from those for other nucleosomes? Furthermore, research
into modulating the nucleosome barrier in vivo is beginning to
converge into a more dynamic view of nucleosomes, rather than viewing them
as static packaging units, which raises the question of how the various
mechanisms for modulating the barrier contribute to overall dynamics of
nucleosomes” (Teves, Weber and Henikoff 2014).
- Several histone chaperone proteins see to the transcription-dependent displacement of histones. Other proteins are thought to reestablish the histones behind the transcribing enzyme, thus reconstituting the nucleosomes and chromatin structure. Lack of the chaperones “results in aberrant transcription from cryptic start sites within transcribed coding regions” (Bell, Tiwari, Thomä and Schübeler 2011).
- “One of the best studied mechanisms for modulating the barrier acts by altering the histone-DNA contacts within the nucleosome through post-translational modification of histones. Much of the research has focused on [histone] H3 modifications and their strong correlation with transcription, and recent reviews provide extensive coverage of the potential role of these modifications in transcription. However, in recent years, mono-ubiquitylation of H2B (H2Bub1) has emerged as a major yet understated player in modulating the nucleosome barrier ... Evidence suggests that “H2Bub1 aids Pol II elongation by stimulating nucleosome remodeling ahead of Pol II and facilitating nucleosome reassembly behind Pol II” (Teves, Weber and Henikoff 2014).
- Histone variants, deposited into nucleosomes, also play a role in facilitating the passage of the transcribing enzyme (RNA polymerase II) along a gene. For example, “the emerging role of [histone variant] H2A.Z in facilitating Pol II transit is to increase accessibility of nucleosomal DNA through dynamic turnover of H2A.Z – H2B dimers”. Likewise, histone variant H3.3 seems to aid the transit of Pol II, but the means by which it does so is not clear (Teves, Weber and Henikoff 2014).
- The structure and dynamics of DNA also appear to play a role in nucleosome structure and stability. “During transcription, the melting [strand separation] of promoter DNA and subsequent translocation of the Pol II machinery generates bidirectional torsional forces: positive torsion ahead of and negative torsion behind the elongating Pol II”. That is, the two DNA strands get more tightly wound around each other ahead of Pol II and more loosely wound behind it. Studies “suggest that transcription-generated torsional stress destabilizes nucleosomes ahead of Pol II to facilitate elongation and promotes nucleosome reassembly behind to maintain chromatin integrity” (Teves, Weber and Henikoff 2014).
-
Memory
“The concept of epigenetic memory was first reported in inflammatory
contexts. Here, we found that additional memories are acquired and
maintained in the chromatin of tissue stem cells, which suggests that stem
cells can store epigenetic memories from different experiences they
encounter. By influencing a stem cell’s responses to future assaults, such
memories can have an impact on long-term tissue fitness in either beneficial
or detrimental ways, depending on context”
(Gonzales, Polak, Matos et al. 2021, doi:10.1126/science.abh2444).
“We establish the existence of epigenetic memory of transcriptional
activation for genes that can be silenced by the Polycomb group. This
property emerges during cell differentiation and allows genes to be stably
switched after a transient transcriptional stimulus. This transcriptional
memory state at Polycomb targets operates in cis; however, rather
than relying solely on read-and-write propagation of histone modifications,
the memory is also linked to the strength of activating inputs opposing
Polycomb proteins, and therefore varies with the cellular context. Our data
and computational simulations suggest a model whereby transcriptional memory
arises from double-negative feedback between Polycomb-mediated silencing and
active transcription. Transcriptional memory at Polycomb targets thus
depends not only on histone modifications but also on the gene-regulatory
network and underlying identity of a cell”
(Holoch, Wassef, Lövkvist et al. 2021, doi:10.1038/s41588-021-00964-2).
-
Nucleosome remodeling
“Chromatin‐associated enzymes are responsible for the installation, removal
and reading of precise post‐translation modifications on DNA and histone
proteins. They are specifically recruited to the target gene by associated
factors, and as a result of their activity, they contribute in modulating
cell identity and differentiation ... DNA, histone tails and histone
surfaces can each function as distinct yet functionally interconnected
anchoring points promoting nucleosome binding and modification”. Regarding
the “many histone modifiers and related readers ... the overarching
conclusion is that besides acting on the same substrate (the nucleosome),
each system functions through characteristic modes of action, which bring
about specific biological functions in gene expression regulation”. “The
emerging notion is that intricate domain and subunit compositions, often
involving both readers and modifiers, make each individual enzymatic system
capable of selectively recognizing nucleosomal particles, depending on their
patterns of histone modifications, DNA accessibility, association with other
co‐repressors and co‐activators and localization within chromatin”
(Speranzini, Pilotto, Sixma and Mattevi 2016, doi:10.15252/embj.201593377).
“The epigenome is sensitive to the availability of metabolites that serve as
substrates of chromatin-modifying enzymes. Links between acetyl-CoA
metabolism, histone acetylation, and gene regulation have been documented,
although how specificity in gene regulation is achieved by a metabolite has
been challenging to answer. Recent studies suggest that acetyl-CoA
metabolism is tightly regulated both spatially and temporally to elicit
responses to nutrient availability and signaling cues. Here we discuss
evidence that acetyl-CoA production is differentially regulated in the
nucleus and cytosol of mammalian cells. Recent findings indicate that
acetyl-CoA availability for site-specific histone acetylation is influenced
through post-translational modification of acetyl-CoA-producing enzymes, as
well as through dynamic regulation of the nuclear localization and chromatin
recruitment of these enzymes”. “Acetyl-CoA production has been shown to
modulate transcriptional responses in various conditions and cell types”
(Sivanand, Viney and Wellen 2017, doi:10.1016/j.tibs.2017.11.004).
Interplay of multiple factors: “We used intestinal stem cells (ISCs)
as a model system to reveal the epigenetic changes coordinating gene
expression programs during [stem cell specification and differentiation]. We
found that two distinct epigenetic mechanisms participate in establishing
the transcriptional program promoting ISC specification from embryonic
progenitors. A large number of adult ISC signature genes are targets of
repressive DNA methylation in embryonic intestinal epithelial progenitors.
On the other hand, genes essential for embryonic development acquire
H3K27me3 and are silenced during ISC specification. We also show that the
repression of ISC signature genes as well as the activation of enterocyte
specific genes is accompanied by a global loss of H2A.Z during ISCs
differentiation. Our results reveal that, already during ISC specification,
an extensive remodeling of chromatin both at promoters and distal regulatory
elements organizes transcriptional landscapes operating in differentiated
enterocytes, thus explaining similar chromatin modification patterns in the
adult gut epithelium”
(Kazakevych, Sayols, Messner et al. 2017, doi:10.1093/nar/gkx167).
“Histones, the fundamental substrates for chromatin-modifying and
remodelling enzymes, are mutated in tumours including gliomas, sarcomas,
head and neck cancers, and carcinosarcomas. Classical ‘oncohistone’
mutations occur in the N-terminal tail of histone H3 and affect the function
of polycomb repressor complexes 1 and 2 (PRC1 and PRC2). ... Here we show
that somatic histone mutations occur in approximately 4% (at a conservative
estimate) of diverse tumour types and in crucial regions of histone
proteins. Mutations occur in all four core histones, in both the N-terminal
tails and globular histone fold domains, and at or near residues that
contain important post-translational modifications”
(Nacev, Feng, Bagert et al. 2019, doi:10.1038/s41586-019-1038-1).
“In contrast to earlier views of nucleosome arrays as uniformly regular
and folded, recent findings reveal heterogeneous array organization and
diverse modes of folding. Local structure variations reflect a continuum of
functional states characterized by differences in post-translational histone
modifications, associated chromatin-interacting proteins and
nucleosome-remodeling enzymes”
(Baldi, Korber and Becker 2020, doi:10.1038/s41594-019-0368-x).
The following is a bit lengthy and heavy, but we need an appreciation — if
only based on a bare glimpse — for how complex all this regulatory action
is:
“Acetylation and butyrylation differ only in chain length, with two and four carbon atoms, respectively. Both PTMs were initially identified in histones, where they substantially overlap. Lysine acetylation and chemically related modifications can influence gene expression simply by neutralizing the positive lysine charge, which loosens the DNA interaction with histones, making it more accessible to transcriptional machinery. In that respect, acetylation and butyrylation should be functionally equivalent, as they are both uncharged. However, histones modified with acetyl or related groups can also be recognized by chromatin-remodeling proteins through specialized domains, such as the bromodomain. In bromodomains, the acyl group is accommodated within a hydrophobic binding pocket and kept in place through interactions between the carbonyl group and an asparagine ‘anchor’. Additionally, a ‘gatekeeper’ residue within the binding pocket restricts the size of the acyl group that can fit in the pocket. For some bromodomains a less bulky gatekeeper allows binding of butyryl chains, while others bind specifically acetyl groups. This allows cells to distinguish these two similar PTMs and to respond differently. For instance, during spermatogenesis, histones become hyper-acetylated and then replaced by transition proteins. The bromodomain-containing protein Brdt is indispensable for this process. Competing butyrylation might control the timing of histone replacement. In fact, Brdt binds less efficiently to butyrylated histones and, as a consequence, histones with this modification are removed later as compared to the acetylated ones.
Crotonylation differs slightly from acetylation and butyrylation, in that a double bond confers a uniquely planar orientation. As for butyrylation, substantial overlap exists between acetylation and crotonylation sites on histones. Only one bromodomain that binds butyrylated peptides also recognizes crotonylation. This is remarkable considering that butyrylation and crotonylation differ only by the double bond, and even antibodies cannot reliably discriminate between the two. YEATS domains have been identified as specific crotonyllysine ‘‘readers.’’ The planar orientation of the crotonyl chain allows it to slide into the YEATS binding site and to engage in a p-p-p stack with two aromatic sidechains so that crotonylated peptides are bound more efficiently than acetylated ones. Hence, cells have the necessary equipment to sense specifically crotonylation. In the yeast metabolic cycle, the phase of high energy availability coincides with a peak in histone acetylation and expression of pro-growth genes, while a peak of histone crotonylation is observed as cells enter the more quiescent phase. The switch from histone acetylation to crotonylation is crucial for turning off pro-growth genes and this involves crotonyllysine sensing by the YEATS-domain-containing protein Taf14” (Figlia, Willnow and Teleman 2020, doi:10.1016/j.devcel.2020.06.036).
“Histone post-translational modifications (PTMs) have emerged as exciting
mechanisms of biological regulation, impacting pathways related to cancer,
immunity, brain function, and more. Over the past decade alone, several
histone PTMs have been discovered, including acylation, lipidation,
monoaminylation, and glycation, many of which appear to have crucial roles
in nucleosome stability and transcriptional regulation”. “New studies
reveal a class of nonenzymatic histone PTMs derived from covalent binding of
highly reactive species, refuting the common notion that histone PTMs
require writers, readers, and erasers for biological significance”
(Chan and Maze 2020, doi:10.1016/j.tibs.2020.05.009).
“Pioneer transcription factors have the ability to access DNA in compacted
chromatin. Multiple transcription factors can bind together to a regulatory
element in a cooperative way, and cooperation between the pioneer
transcription factors OCT4 (also known as POU5F1) and SOX2 is important for
pluripotency and reprogramming. However, the molecular mechanisms by which
pioneer transcription factors function and cooperate on chromatin remain
unclear. Here we present cryo-electron microscopy structures of human OCT4
bound to a nucleosome containing human LIN28B or nMATN1 DNA sequences, both
of which bear multiple binding sites for OCT4. Our structural and
biochemistry data reveal that binding of OCT4 induces changes to the
nucleosome structure, repositions the nucleosomal DNA and facilitates
cooperative binding of additional OCT4 and of SOX2 to their internal binding
sites. The flexible activation domain of OCT4 contacts the N-terminal tail
of histone H4, altering its conformation and thus promoting chromatin
decompaction. Moreover, the DNA-binding domain of OCT4 engages with the
N-terminal tail of histone H3, and post-translational modifications at H3K27
modulate DNA positioning and affect transcription factor cooperativity.
Thus, our findings suggest that the epigenetic landscape could regulate OCT4
activity to ensure proper cell programming”
(Sinha, Bilokapic, Du et al. 2023; doi:10.1038/s41586-023-06112-6).
“The precise effect of chromatin modifications is influenced by multiple
contextual factors, including the underlying DNA sequence, transcription
factor occupancy and genomic positioning”
(Anonymous 2024, doi:10.1038/s41588-024-01705-x).
“Gene transcription is intimately linked to chromatin state and histone
modifications. However, the enzymes mediating these post-translational
modifications have many additional, nonhistone substrates, making it
difficult to ascribe the most relevant modification”
(Mannervik 2024, doi:10.1101/gad.351969.124). In other words, gene
regulation is so thoroughly wrapped up with everything else going on in the
cell that it is hard to disentangle gene regulation from “everything”.
-
Histone tail modifications
The core histones of nucleosomes have flexible, filamentary “tails”.
Numerous distinct modifications of these tails have been identified
(often called “marks”). These involve the placement of any one of a
considerable number of chemical groups on particular amino acid
residues of the tails. This can alter the charge on the histone or
else provide binding sites for regulatory proteins. Either way, the
modifications can directly affect gene expression, and can also affect
expression indirectly by helping to determine the structure of
chromatin. The effects depend on (1) which chemical group is involved;
(2) which amino acid on which histone tail the chemical group attaches
to; (3) where in relation to a gene the affected nucleosome is located;
(4) particularly in the case of methyl groups, whether one, two, or
three copies of the group are attached to the amino acid; and (5) the
larger context, and in particular, the context of other nearby
modifications. It is impossible to summarize here all the (more or
less approximate) patterns of modification that have been found
significant for one or another aspect of gene expression. There are
combinatorial possibilities here that rival those of the genome itself.
The “incredible diversity of histone modifications leads naturally to
the question of what it all means — why do so many histone
modifications occur in the cell? this question only becomes more
vexing when considering that even in the past year mass spectrometry
studies have identified scores of previously unknown histone
modifications” (Rando 2012).
“At the level of the primary chromatin structure, the data suggest
that [maps of] histone modifications indicate functional genomic
elements, gene expression, splicing patterns and modes of repression.
... Additionally, these maps promote an appreciation of the
three-dimensional organization of the genome. ... Histone modifications
are intimately tied to large-scale repressive domains like LADs
[lamina-associated domains] and Polycomb bodies” (Zhou, Goren and
Bernstein 2010).
“Histone modifications are linked to essentially every cellular process
requiring DNA access, including transcription, replication and repair”.
Recent studies “point to a view of histone modifications as cogs in
dynamic chromatin processes, wherein histone modifications reinforce
changes in nucleosome occupancy, positioning or composition mediated by
processes such as transcriptional elongation, chromatin remodeling and
the targeting actions of noncoding RNAs” (Zentner and Henikoff 2013).
“A review of the recent literature reveals that novel sites or types of
histone PTMs are rapidly being discovered and characterized ... The
diversity seen in terms of location on the nucleosome, genome
localization and the cellular processes in which they are involved
highlight the importance of histone PTMs to multiple fields of study
including cell biology, epigenetics, development and cancer biology.
... The sheer number of novel modifications begs the question how many
more types of PTMs are there remaining to be found?” (Arnaudo and
Garcia 2013).
Histone tail modifications influence “all DNA-based processes, including
chromatin compaction, nucleosome dynamics, and transcription”
(Lawrence, Daujat and Schneider 2016, doi:10.1016/j.tig.2015.10.007).
“Modifications affecting the globular histone core have been uncovered as
being crucial for DNA repair, pluripotency and oncogenesis”
(Tessadori, Giltay, Hurst et al. 2017, doi:10.1038/ng.3956).
“Small histone modifications (acetylation, methylation) can alter
nucleosome charge and/or subtly affect nucleosome dynamics. Here,
Krajewski argues that when the chemical modification approaches the size
of a histone, such as ubiquitylation and SUMOylation, gross structural
distortions and large-scale changes in nucleosome dynamics occur ...
“Regulated installation of large histone modifications is associated with DNA-dependent processes including transcription and replication. Krajewski suggests that this added bulk may trigger spontaneous, transient, and reversible increases in histone dynamics allowing DNA translocating enzymes to traverse nucleosomes more easily. The potential to distort native structure of canonical nucleosomes may expose intermediate nucleosome structures that can be specifically recognized by nucleosome-interacting proteins. Minor nucleosome instabilities resulting from smaller histone modifications may accelerate deposition of bulky histone modifications through allosteric effects. A tunable range of nucleosome dynamics that crescendos with the addition of bulky modifications may be written within the histone code” (Orlandi and McKnight 2019, doi:10.1002/bies.201900217).
“For many decades research on histone modifications has been focused
almost solely on the biological role of modifications occurring at the
side-chain of internal amino acid residues [that is, histone
modifications that occur “internally” — on the tails or core histones,
but not at the termini of the tails]. In contrast, modifications on the
terminal N-alpha amino group of histones—despite being highly abundant
and evolutionarily conserved—have been largely overlooked. This oversight
has been due to the fact that these marks were being considered inert
until recently, serving no regulatory functions. However, during the past
few years accumulating evidence has drawn attention towards the
importance of chemical marks added at the very N-terminal tip of histones
and unveiled their role in key biological processes including aging and
carcinogenesis. Further elucidation of the molecular mechanisms through
which these modifications are regulated and by which they act to
influence chromatin dynamics and DNA-based processes like transcription
is expected to enlighten our understanding of their emerging role in
controlling cellular physiology and contribution to human disease”
(Demetriadou, Koufaris and Kermizis 2020, doi:10.1186/s13072-020-00352-w).
“In the case of histone Nt-ac [N-alpha terminal acetylation], this CTM
[co-translational modification]has in recent years been implicated in
transcriptional activation, conveying environmental signals and
controlling the expression of specific genetic pathways. A fundamental
mechanism through which Internal PTMs define gene expression patterns is
by cross-regulating each other. The work of different groups, which
is summarized in this review, strongly supports that Nt-modifications
control transcription through their cross-talk with distinct In-marks
[internal modifications]”
(Demetriadou, Koufaris and Kermizis 2020, doi:10.1186/s13072-020-00352-w).
-
General considerations
- Efforts to define a fixed “code” specifying the meaning of particular marks or their combinations have been troubled by ongoing findings. “The greater the resolution and percentage of the genome that is covered by epigenomics, the more these canonical associations between a given mark and gene expression become nuanced and idiosyncratic” (Ruthenburg, Li, Patel and Allis 2007). “One histone modification can influence the reading or writing of another in many different ways” (Justin, De Marco, Aasland and Gamblin 2010).
- Many regulatory proteins can recognize specific histone tail modifications and bind to the DNA or chromatin at those sites. A single protein often responds with loose definition to multiple marks or contexts, and a single mark may attract multiple proteins (more than 10 proteins are known to bind the mark known as H3K4me3).
- Histone modification (particularly H3K4me3) has been found to identify alternative promoters (Pal, Gupta, Kim et al. 2011). (See “Promoters” above and also “Alternative coding sequences (transcription start and termination)” below.)
- In a study of eight different ATP-dependent chromatin remodelers in mouse embryonic stem cells: “Two trends emerge: an activating remodeller in one class of genes is an inhibitor remodeller in the other class; and within the same class, an activating remodeller can be counteracted by an inhibitor remodeller. Taken together, remodellers work together at specific nucleosome positions adjacent to promoter region NFRs [nucleosome-free regions] to elicit proper gene control” (Dieuleveult, Yen, Hmitou et al. 2016, doi:10.1038/nature16505).
- Further information about the foregoing item: “Surprisingly, large CpG-rich NFRs that extend downstream of annotated transcriptional start sites are nevertheless bound by non-nucleosomal or subnucleosomal histone variants (H3.3 and H2A.Z) and marked by H3K4me3 and H3K27ac modifications. RNA polymerase II therefore navigates hundreds of base pairs of altered chromatin in the sense direction before encountering [the bounding, canonical] nucleosome at the 3′ end of the NFR. Transcriptome analysis after remodeller depletion reveals reciprocal mechanisms of transcriptional regulation by remodellers. Whereas at active genes individual remodellers have either positive or negative roles via altering nucleosome stability, at polycomb-enriched bivalent genes the same remodellers act in an opposite manner. These findings indicate that remodellers target specific nucleosomes at the edge of NFRs, where they regulate ES cell transcriptional programs” (Dieuleveult, Yen, Hmitou et al. 2016, doi:10.1038/nature16505).
- The chemical groups constituting the marks include the following, among others. (To document the distinct yet interwoven roles of these modifications would require a huge amount of space, and also a lot of editing over time, since the picture is continually being revised and expanded. I mention a few examples more or less at random.)
-
Some particular modifications
Histone tail modifications are so numerous, and their significances
have been, and are being, so extensively traced, that I am no longer
making much of an effort to keep up with developments. There are few
aspects of gene regulation that do not intersect, in one way or
another, with histone tail modifications, of which the combinatorial
possibilities seem almost infinite.
-
Methylation
Note that this is not the
DNA methylation described above.
- Methylation of certain amino acids on certain histone tails in certain locations with respect to a gene’s start-site is associated with active transcription or transcription-readiness. Other methylations are associated with gene silencing. But this is simplistic. For example, an activating methylation can coexist with a repressive mark on the same histone tail in stem cells, leading to what is called a “bivalent” or “poised” state. Such combinations, it is thought, helps to maintain developmental genes in a condition where they can be quickly activated when the cell is ready to commit to differentiation.
- Most attention has been given to the methylation of various lysine residues on the histone tails. These residues can be mono-, di-, or tri-methylated. However, other residues can also be methylated, such as arginine. Interestingly, H3R2 (arginine as the second residue of the N-terminal tail of histone H3) can be di-methylated in two ways: asymmetrically or symmetrically — with very different effects. Asymmetric di-methylation tends to be repressive and antagonistic to normally activating H3K4 tri-methylation. But symmetric di-methylation of H3R2 corresponds to a highly expressed form of chromatin (“euchromatin”), “revealing that subtle steric changes at this site can result in markedly different molecular and functional consequences for transcriptional regulation” (Migliori, Müller, Phalke et al. 2012).
- A wholly different kind of symmetry or asymmetry involves the paired histones in the canonical core histone octamer, which consists of two each of four different histones. The significance of a particular tail modification can depend on whether the two tails of a histone pair are symmetrically or asymmetrically modified — that is, on whether just one or both tails have the modification. For example, “Polycomb repressive complex 2-mediated methylation of H3K27 was inhibited when nucleosomes contain symmetrically, but not asymmetrically, placed H3K4me3 or H3K36me3” (Voigt, LeRoy, Drury et al. 2012).
- Mouse olfactory neurons contain more than 1000 genes for odorant receptors, but the mystery has been how it can be that each neuron expresses only one of those genes. Management of the chromatin state by the histone demethylase LSD1 appears to be the key: “LSD1 (in complex with a yet-unknown H3K9me3 demethylase) chooses a single OR allele by reversing its previously heterochromatinized state and facilitating the acquisition at the allele of a transcriptionally active H3K4me3 signature. If the chosen allele encodes a functional odorant receptor (OR), expression of Adenylyl Cyclase 3 is induced and results in the downregulation of LSD1, thereby preventing activation of other OR gene alleles. The authors refer to this activation of a single allele and prevention of activation of other alleles as an ‘epigenetic trap’ that locks in the singular choice of one allele of one OR gene” (Reinsborough and Chess reporting on work by Lyons, Allen, Goh et al. 2013).
- In a study of Caenorhabditis elegans: “H3K9 methylation (K9me) is enriched in repetitive elements (REs) and suppresses repetitive element transcription. In the absence of the methyltransferases required for H3K9 methylation (met-25 and met-2), H3K9 methylation is lost and repetitive elements are aberrantly transcribed. Unscheduled transcription of repetitive elements leads to R-loop formation and mutations specifically at the deregulated repetitive elements” (Salcini 2016, doi:10.1038/ng.3705).
- “It has long been observed that hypoxia induces histone lysine hypermethylation, a form of epigenetic chromatin modification. However, whether this represents a direct sensing of oxygen tension or an indirect effect ... has not been established. [New evidence, however, demonstrates] in different cellular systems that the activity of the lysine-specific demethylases (KDMs) KDM5A and KDM6A is oxygen sensitive, and thereby identifying them as oxygen sensors” (Gallipoli and Huntly 2019, doi:10.1126/science.aaw8373).
- “Choudhury et al. report that yeast Set1C/COMPASS is dimeric and, consequently, symmetrically trimethylates histone 3 Lys4 (H3K4me3) on promoter nucleosomes. This presents a new paradigm for the establishment of epigenetic detail, in which dimeric methyltransferase and monomeric demethylase cooperate to eliminate asymmetry and focus symmetrical H3K4me3 onto selected nucleosomes” (TOC blurb for Choudhury, Singh, Arumugam2 et al. 2019, doi:10.1101/gad.322222.118).
- “Epigenetic mechanisms contribute to the regulation of cell differentiation and function. Vascular smooth muscle cells (SMCs) are specialized contractile cells that retain phenotypic plasticity even after differentiation. Here, by performing selective demethylation of histone H3 lysine 4 di-methylation (H3K4me2) at SMC-specific genes, we uncovered that H3K4me2 governs SMC lineage identity. Removal of H3K4me2 via selective editing in cultured vascular SMCs and in murine arterial vasculature led to loss of differentiation and reduced contractility due to impaired recruitment of the DNA methylcytosine dioxygenase TET2. H3K4me2 editing altered SMC adaptative capacities during vascular remodeling due to loss of miR-145 expression. Finally, H3K4me2 editing induced a profound alteration of SMC lineage identity by redistributing H3K4me2 toward genes associated with stemness and developmental programs, thus exacerbating plasticity. Our studies identify the H3K4me2-TET2-miR145 axis as a central epigenetic memory mechanism controlling cell identity and function, whose alteration could contribute to various pathophysiological processes” (Liu, Espinosa-Diez, Mahan et al. 2021, doi:10.1016/j.devcel.2021.09.001).
- “Ash1 is a histone H3K36 methyltransferase and is involved in gene activation. Ash1 forms a large complex with Mrg15 and Caf1/p55/Nurf55/RbAp48 (AMC complex). The Ash1 subunit alone exhibits very low activity due to the autoinhibition, and the binding of Mrg15 releases the autoinhibition. Caf1 is a scaffolding protein commonly found in several chromatin modifying complexes and has two histone binding pockets: one for H3 and the other for H4. Caf1 has the ability to sense unmodified histone H3K4 residues using the H3 binding pocket ... Here, we dissected the interaction among the AMC complex subunits, revealing that Caf1 uses the histone H4 binding pocket to interact with Ash1 near the histone binding module cluster. Furthermore, we showed that H3K4 methylation inhibits AMC HMTase activity via Caf1 sensing unmodified histone H3K4 to regulate the activity in an internucleosomal manner, suggesting that crosstalk between H3K4 and H3K36 methylation. Our work revealed a delicate mechanism by which the AMC histone H3K36 methyltransferase complex is regulated” (Yoon and Song 2023, doi:10.1186/s13072-023-00487-6).
-
Acetylation
“Histone acetylation and non-histone protein acetylation influence
a myriad of cellular and physiological processes, including
transcription, phase separation, autophagy, mitosis,
differentiation and neural function. The activity of lysine
acetyltransferases and lysine deacetylases can, in turn, be
regulated by metabolic states, diet and specific small molecules.
Histone acetylation has also recently been shown to mediate
cellular memory. These features enable acetylation to integrate the
cellular state with transcriptional output and cell-fate decisions”
(Shvedunova and Akhtar 2022, doi:10.1038/s41580-021-00441-y).
-
Acetylation of various histone tail locations is generally
associated with transcription-readiness. More
particularly, it facilitates decompaction of chromatin, the
loosening of contacts between DNA and histones, and
interaction between histones and various regulatory
proteins.
- Not only protein-coding genes can be activated. For example, histone acetylation, along with DNA demethylation, activates expression of an miRNA, resulting in apoptosis of gastric cancer cells (Saito, Suzuki, Tsugawa et al. 2009).
- Hypoacetylation (loss of acetylation) has been generally associated with gene silencing. However, “histone deacetylases [which remove acetyl groups] have now also been found to be abundantly present on active genes in human cells” (Steensel 2011). It may be that there are highly dynamic processes going on, involving well-timed application and removal of acetyl groups.
- “Our single-cell analysis reveals histone H3 lysine-27 acetylation at a gene locus can alter downstream transcription kinetics by as much as 50%, affecting two temporally separate events. First acetylation enhances the search kinetics of transcriptional activators, and later the acetylation accelerates the transition of RNAP2 [RNA polymerase II) from initiation to elongation. Signatures of the latter can be found genome-wide using chromatin immunoprecipitation followed by sequencing. We argue that this regulation leads to a robust and potentially tunable transcriptional response” (doi:10.1038/nature13714).
- “Comprehensive benchmarking reveals H2BK20 acetylation as a distinctive signature of cell-state-specific enhancers and promoters” (article title: Kumar, Rayan, Muratani et al. 2016, doi:10.1101/gr.201038.115).
- Beyond acetylation: “In addition to acetylation, eight types of structurally and functionally different short-chain acylations have recently been identified as important histone Lysine modifications: propionylation, butyrylation, 2-hydroxyisobutyrylation, succinylation, malonylation, glutarylation, crotonylation [see also below] and β-hydroxybutyrylation. These modifications are regulated by enzymatic and metabolic mechanisms and have physiological functions, which include signal-dependent gene activation and metabolic stress”. The physiological functions of non-acetyl acylation also include spermatogenesis and tissue injury response. “Differential histone acylation is regulated by the metabolism of the different acyl-CoA forms, which in turn modulates the regulation of gene expression” (Sabari, Zhang, Allis and Zhao 2017,doi:10.1038/nrm.2016.140).
-
Acetylation of various histone tail locations is generally
associated with transcription-readiness. More
particularly, it facilitates decompaction of chromatin, the
loosening of contacts between DNA and histones, and
interaction between histones and various regulatory
proteins.
-
Phosphorylation
- As one example of histone phosphorylation: phosphorylation of the serine 47 residue of histone H4 promotes nucleosome assembly that brings together phosphorylated H4 with the variant histone, H3.3 rather than the canonical histone H3.1. H3.3 has been found enriched in the bodies of actively transcribed genes, and also plays a role in heterochromatin formation. In mice, loss of function of the H3.3 histone commonly results in postnatal death and male infertility (Kang, Pu, Hu et al. 2011).
- One other example: “Although histone H3 phosphorylation is a target of numerous signaling pathways, its role in transcriptional regulation remains poorly understood ... We report a genome-wide analysis of H3S28 phosphorylation in a mammalian system in the context of stress signaling. We found that this mark targets as many as 50% of all stress-induced genes, underlining its importance in signal-induced transcription ... We found that MSK1/2-mediated phosphorylation of H3S28 at stress-responsive promoters contributes to the dissociation of HDAC [histone deacetylase] corepressor complexes and thereby to enhanced local histone acetylation and subsequent transcriptional activation of stress-induced genes” (doi:10.1101/gr.176255.114).
- “Phosphorylation of histone H3.3 at serine 31 by CHK1 is shown to stimulate activity of the acetyltransferase p300 in trans. Depletion of histone H3.3 in embryonic stem cells reduces enhancer acetylation during differentiation” (toc blurb for Martire, Gogate, Whitmill et al. 2019, doi:10.1038/s41588-019-0428-5).
-
Ubiquitination (or “ubiquitylation”)
Ubiquitin is a small protein that various enzymes apply to many
of the body’s proteins, including histones, as
post-translational modifications. “(1) Proteins can be
modified with a single ubiquitin or with polymeric chains that
differ in the connection between ubiquitin molecules. (2) The
different ubiquitin modifications adopt distinct structures.
(3) Ubiquitin-binding proteins exploit various strategies to
specifically interact with particular types of ubiquitin
modifications. (4) Ubiquitin chains can be disassembled by
nonspecific or linkage-specific deubiquitinating enzymes. (5)
The various ubiquitin modifications trigger a wide range of
biological reactions, including protein degradation,
activation, and localization. (6) The consequences of
ubiquitylation are determined by the chain topology in
combination with additional factors, such as substrate
localization or sensitivity to deubiquitylation” (Komander and
Rape 2012). So far as is currently known, histone tails are
typically only monoubiquitylated. (Transcription factors, on
the other hand, are subject to the full range of
ubiquitin-related modifications.)
- Factors that remove ubiquitin from histone tails are as important as those that add them: “DUBs [deubiquitylating enzymes] are integral components of the transcription machinery, involved in both gene activation and repression. They modulate the ubiquitylation status of histones H2A and H2B, which play pivotal roles in a cascade of molecular events that determine chromatin status. A DUB module in the SAGA coactivator complex is required for gene activation, whereas other DUBs are part of the Polycomb gene-silencing machinery. DUBs also control the level or subcellular compartmentalization of selective transcription factors, including the tumour suppressor p53. Typically, DUB specificity and activity are defined by its partner proteins, enabling remarkably versatile and sophisticated regulation” (Frappier and Verrijzer 2011).
- Rhythm and timing play a role in ubiquitin-related gene regulation: “A temporal cycle of H2B ubiquitylation followed by deubiquitylation is required for optimal gene activation” (Frappier and Verrijzer 2011).
- DUBs (deubiquitylating enzymes) are regulated even as they regulate gene expression. “Gene control by DUBs involves a wide variety of distinct mechanisms. (De)ubiquitylation can control the level or subcellular localization of key transcription factors in response to signaling. Another emerging theme is that associated partner proteins control the activity and specificity of DUBs. Selective DUBs can be targeted to specific genomic loci by transcription factors, sometimes involving cooperative DNA binding. Generally, DUBs appear to be part of extensive protein-interaction networks” (Frappier and Verrijzer 2011).
- “We demonstrate the direct involvement of [human] H2B monoubiquitination in centromeric chromatin maintenance. Monoubiquinated H2B (H2Bub1) is needed for this maintenance, promoting noncoding transcription, centromere integrity and accurate chromosomal segregation. A transient pulse of centromeric H2Bub1 leads to RNA polymerase II–mediated transcription of the centromere’s central domain, coupled to decreased H3 stability. H2Bub1-deficient cells have centromere cores that, despite their intact centromeric heterochromatin barriers, exhibit characteristics of heterochromatin, such as silencing histone modifications, reduced nucleosome turnover and reduced levels of transcription. ... Centromeric H2Bub1 is essential for maintaining active centromeric chromatin” (Sadeghi, Siggens, Svensson and Ekwall 2014).
- The first indication of a connection between the multi-protein Mediator of the pre-initiation complex and post-translational histone modification on active genes: “The Mediator core complex, which is composed of 26 subunits, stabilises promoter/enhancer loops by physically bridging transcription factors bound at enhancer elements with the RNA polymerase II transcription machinery at core promoter regions, thereby coordinating transcription initiation events. The Mediator subunit MED23, either alone or in a specialised mediator complex, associates with the E3‐ligase RNF20/40 to promote the H2BK120ub mark along the gene body of an actively transcribed gene, thereby promoting transcriptional elongation” (Streubel, Adrian P Bracken 2015, doi:10.15252/embj.201592996).
- “Ubiquitination of histone H2B provides an important checkpoint in the transition from the early initiated form of RNA polymerase II to the full elongating form. This change is governed by the phosphorylation status of heptapeptide repeats in the carboxyl-terminal domain (CTD) of the largest subunit of RNA polymerase II. Immediately after initiation, these repeats are phosphorylated on serine 5 and serine 7, which brings cofactors to the polymerase that facilitate early elongation steps. These repeats are then phosphorylated on serine 2, which recruits cofactors that function during subsequent transcription elongation (5). Monoubiquitination of the carboxyl-terminal tail of H2B blocks the enzyme that phosphorylates serine 2 of the CTD repeats, thus regulating the transition to full elongation. Deubiquitination of H2B by the SAGA complex allows phosphorylation of serine 2 of the CTD repeats, promoting transition from the early elongation to the full elongation form of RNA polymerase II” (Workman 2016, doi:10.1126/science.aaf1495).
-
ADP-ribosylation
Histone residues can be reversibly “marked” with single
ADP-ribose moieties, and these can be extended into (possibly
branching) chains of ADP-ribose. Much work is going on now to
discover the functional significance of ADP-ribosylation
patterns.
- “ADP-ribosylation activity is associated primarily with transcriptionally active regions. ... [Experiments indicate] the importance of ADP-ribosylation in processes that involve broad chromatin rearrangements and changes in the transcriptional states of cells” — changes such as those that occur during cell differentiation (Messner and Hottiger 2011).
-
Crotonylation
“An increasing number of studies have demonstrated that histone
crotonylation at DNA regulatory elements plays an important role
in the activation of gene transcription. However, among others,
we have shown that elevated cellular crotonylation levels result
in the inhibition of endocytosis-related gene expression and
pro-growth gene expression, implicating the complexity of
histone crotonylation in gene regulation.”
“Lysine crotonylation (Kcr), a previously unknown
post-translational modification (PTM), plays crucial roles in
regulating cellular processes, including gene expression,
chromatin remodeling, and cellular metabolism. Kcr is associated
with various diseases, including neurodegenerative disorders,
cancer, cardiovascular conditions, and metabolic syndromes”
(Ji, Liu, Zhang et al. 2025, doi:10.1186/s13072-025-00577-7).

Histone crotonylation and some of its regulating factors. The schematic model shows the principal lysine crotonylation sites on histones H3 and H4 and the reported writers (crotonyltransferase), erasers (decrotonylase), readers, and other regulators for each lysine crotonylation.
Credit: Li, Kun and Ziqiang Wang (2021). “Histone Crotonylation-Centric Gene Regulation”, pigenetics and Chromatin vol. 14, no. 10 (February 6). cc by 4.0
“Increasing evidence has demonstrated that histone Kcr [lysine
crotonylation] is associated with physiological and pathological
processes, such as differentiation, tissue injury, virus
infection, tumorigenesis, and neurodegenerative disease. The
first histone Kcr-related biological process to be discovered
was germ cell differentiation. During spermatogenesis, histone
Kcr is enriched in promoters of highly expressed testes genes,
including a number of X-linked genes that function to maintain
sex chromosome activation in haploid cells. This results in the
differentiation of male germinal cells immediately following
meiosis. Histone Kcr was next found to be related to
nephropathy, including acute kidney injury (AKI). Researchers
found that increased histone crotonylation prevented AKI and a
decrease in renal function via increasing PGC-1α and sirtuin-3
levels and decreasing CCL2 expression”
(Li and Wang 2021, doi:10.1186/s13072-021-00385-9).
- In mammals, post-meiotic male germ cells have most of their sex-linked genes repressed. However, a subset is active, and crotonylation of a histone lysine has been found to mark these active genes, apparently conferring resistance to transcriptional repressors. The same histone modification was found on post-meiotically active, testis-specific genes on autosomes (chromosomes other than sex chromosomes) (Montellier, Rousseaux, Zhao and Khochbin 2012).
- “Histone lysine acetylation at DNA regulatory elements promotes transcriptional activation ... Allis and colleagues now report that p300-catalysed histone crotonylation is a more potent transcriptional activator than histone acetylation. They also find that whether histone lysines are crotonylated or acetylated depends on the relative intracellular concentrations of crotonyl-CoA and acetyl-CoA, thereby linking cellular metabolism to gene expression” (Baumann 2015, doi:10.1038/nrm3992; reporting on work by Sabari et al. 2015, doi:10.1016/j.molcel.2015.02.029).
- Hydroxylation. Hydroxylation of tyrosine residues as well as crotonylation (see immediately above) were recently discovered (Tan 2011).
-
Serotonylation.
“Here we provide evidence for a class of histone post-translational
modification, serotonylation of glutamine, which occurs at position
5 (Q5ser) on histone H3 in organisms that produce serotonin (also
known as 5-hydroxytryptamine (5-HT)). We demonstrate that tissue
transglutaminase 2 can serotonylate histone H3 tri-methylated
lysine 4 (H3K4me3)-marked nucleosomes, resulting in the presence of
combinatorial H3K4me3Q5ser in vivo. H3K4me3Q5ser displays a
ubiquitous pattern of tissue expression in mammals, with enrichment
observed in brain and gut, two organ systems responsible for the
bulk of 5-HT production. Genome-wide analyses of human serotonergic
neurons, developing mouse brain and cultured serotonergic cells
indicate that H3K4me3Q5ser nucleosomes are enriched in euchromatin,
are sensitive to cellular differentiation and correlate with
permissive gene expression, phenomena that are linked to the
potentiation of TFIID interactions with H3K4me3. Cells that
ectopically express a H3 mutant that cannot be serotonylated
display significantly altered expression of H3K4me3Q5ser-target
loci, which leads to deficits in differentiation. Taken together,
these data identify a direct role for 5-HT, independent from its
contributions to neurotransmission and cellular signalling, in the
mediation of permissive gene expression” (Farrelly, Thompson et al.
2019, doi:10.1038/s41586-019-1024-7).
“Serotonylation of histones and its potential influence on
transcription might be only the tip of the iceberg in an
ever-expanding scenario of post-translational modifications
associated with chromatin changes. Histaminylation and
dopaminylation (addition of histamine, an amino acid, and dopamine,
a neurotransmitter, respectively) are likely to join the party,
which could complicate the task of deciphering the language of
histone modifications”
(Cervantes and Sassone-Corsi 2019, doi:10.1038/d41586-019-00532-z)
“Serotonylation of H3Q5 is the first endogenous monoaminyl
modification, and the first non-methyl post-translational
modification of Gln, to be identified in histones. H3Q5ser
promotes the transcription of neuronal genes during neuronal cell
differentiation by potentiating the binding of the H3K4me3 reader
TFIID at the gene promoters”
(Zlotorynski 2019, doi:10.1038/s41580-019-0124-4).
Gln and Q both stand for the glutamine amino acid.
- “Astrocytes can respond to input from neuromodulators. However, the roles for neuromodulators in astrocytes in functioning brain circuits are poorly defined. Sardar et al. discovered that loss of the astrocytic neuromodulator transporter Slc22a3 resulted in reduced levels of intracellular serotonin and impaired calcium ion–mediated serotonin responses (see the Perspective by Vasile and Rouach). These deficits in serotonin manifest in astrocytes with a Slc22a3 deletion that have reduced histone serotonylation. The expression of genes for the synthesis of γ-aminobutyric acid (GABA) is regulated by histone serotonylation. These changes in GABA gene expression are accompanied by reduced GABA release from astrocytes that lack Slc22a3. Inhibition of histone serotonylation in olfactory bulb astrocytes leads to reduced GABA release and impaired olfactory sensory processing” (Sardar, Cheng, Woo et al. 2023; doi:10.1126/science.ade00).
-
Sumoylation
“Recent global proteomic and genetic studies have linked
modification by the small ubiquitin-related modifier (SUMO) to
many processes involving chromatin, including transcriptional
activation and repression...” “Posttranslational modification
of [histones and other] proteins by small ubiquitin-related
modifiers (SUMOs) regulates chromatin structure and function at
multiple levels and through a variety of mechanisms to
influence gene expression and maintain genome integrity”.
“Sumoylation modulates gene expression through effects on DNA
methylation, histones, and transcriptional regulators”
(Cubeñas-Potts and Matunis 2013).
“A recent study identifies SUMOylation as a guardian of cell
identity that acts during differentiation and reprogramming by
reinforcing active enhancers and maintaining silenced
heterochromatin in a context-specific manner.” “Observation that
SUMO safeguards cell identity in a context-specific manner by either
reinforcing active enhancers in MEFs [mouse embryonic fibroblasts]
or silenced heterochromatin in ESCs [embryonic stem cells] reveals
the unexpected versatility of this PTM in positively or negatively
modulating gene expression”. In particular: “(A) Hyposumoylation
facilitates the reprogramming of mouse embryonic fibroblasts (MEFs)
into induced pluripotent stem cells (iPSCs) and the conversion of
ESCs to 2C- [two-cell blastomere]-like cells. (B) SUMO suppression
facilitates cell fate change in different cellular systems with or
without ectopic expression of transcription factors (TFs). (C) SUMO
influences enhancer activity and heterochromatin levels. In MEFs,
SUMO facilitates the cooperative binding of somatic TFs at
enhancers. In ESCs, SUMOylation of components of the PRC1.6 complex
and of heterochromatic factors such as SETDB1, HP1a, and KAP1
contributes to the silencing of endogenous retroviral
element-associated genes”
(Di Stefano and Hochedlinger 2018, doi:10.1016/j.tcb.2018.10.001).
“It was initially thought that histone sumoylation exclusively
suppressed gene transcription, but recent advances in proteomics
and genomics have uncovered its diverse functions in
cotranscriptional processes, including chromatin remodeling,
transcript elongation, and blocking cryptic initiation. Histone
sumoylation is integral to complex signaling codes that prime
additional histone PTMs as well as modifications of the RNA
polymerase II carboxy-terminal domain (RNAPII-CTD) during
transcription. In addition, sumoylation of histone variants is
critical for the DNA double-strand break (DSB) response and for
chromosome segregation during mitosis”
(Ryu and Hochstrasser 2021, doi:10.1093/nar/gkab280).
- “Sumoylation often functions as a signal to facilitate protein-protein interactions on chromatin. These interactions may be simple heterodimeric associations, but they can also involve very large multiprotein complexes” (Cubeñas-Potts and Matunis 2013).
- “Sumoylation also specifies multiple other fates, including effects on enzyme activity and change in protein subcellular localization” (Cubeñas-Potts and Matunis 2013).
- “Although in many cases sumoylation is linked to heterochromatin and gene inactivation, a growing number of studies indicate that sumoylation also plays important roles in enhancing chromatin accessibility and gene activation. Thus, the effects of sumoylation are dichotomous and often context dependent” (Cubeñas-Potts and Matunis 2013).
- “We found that, whereas SUMO alone is widely distributed over the genome with strong association at active promoters, active sumoylation occurs most prominently at promoters of histone and protein biogenesis genes, as well as Pol I rRNAs and Pol III tRNAs. Remarkably, these four classes of genes are up-regulated by inhibition of sumoylation, indicating that SUMO normally acts to restrain their expression. In line with this finding, sumoylation-deficient cells show an increase in both cell size and global protein levels. Strikingly, we found that in senescent cells, the SUMO machinery is selectively retained at histone and tRNA gene clusters, whereas it is massively released from all other unique chromatin regions. These data ... reveal the highly dynamic nature of the SUMO landscape” (Neyret-Kahn, Benhamed, Ye et al. 2013).
- “SUMO homeostasis is important for many cellular processes ... Liang and colleagues demonstrate how a desumoylation enzyme is targeted to the nucleolus for removing SUMO from specific substrates and how curtailing sumoylation levels can regulate transcription in this nuclear compartment” (Dhingra and Zhao 2017, doi:10.1101/gad.300491.117).
- “Here, we identify the protein network around chromatin-bound glucocorticoid receptor [GR] by using selective isolation of chromatin-associated proteins and show that the network is affected by receptor SUMOylation, with several nuclear receptor coregulators and chromatin modifiers preferring interaction with SUMOylation-deficient GR and proteins implicated in transcriptional repression preferring interaction with SUMOylation-competent GR ... the SUMOylation-deficient GR is more potent in binding and opening chromatin at glucocorticoid-regulated enhancers and inducing expression of target loci ... Our results thus show that SUMOylation modulates the specificity of GR by regulating its chromatin protein network and accessibility at GR-bound enhancers. We speculate that many other SUMOylated TFs utilize a similar regulatory mechanism” (Paakinaho, Lempiäinen, Sigismondo, Niskanen et al. 2021, doi:10.1093/nar/gkab032).
-
O-GlcNAcylation
This is a nutrient-sensitive sugar modification that, applied
to more than just histones, participates in the epigenetic
regulation of gene expression. The enzymes applying this
modification “target key transcriptional and epigenetic
regulators including RNA polymerase II, histones, histone
deacetylase complexes and members of the Polycomb and Trithorax
groups. Thus, O‑GlcNAc cycling may serve as a
homeostatic mechanism linking nutrient availability to
higher-order chromatin organization. In response to nutrient
availability, O‑GlcNAcylation is poised to influence X
chromosome inactivation and genetic imprinting, as well as
embryonic development. The wide range of physiological
functions regulated by O‑GlcNAc cycling suggests an
unexplored nexus between epigenetic regulation in disease and
nutrient availability” (Hanover, Krause and Love 2012).
- “The glycosyltransferase Ogt adds O-linked N-Acetylglucosamine (O-GlcNAc) moieties to nuclear and cytosolic proteins. Drosophila embryos lacking Ogt protein arrest development with a remarkably specific Polycomb phenotype, arising from the failure to repress Polycomb target genes. The Polycomb protein Polyhomeotic (Ph), an Ogt substrate, forms large aggregates in the absence of O-GlcNAcylation both in vivo and in vitro. O-GlcNAcylation of a serine/threonine (S/T) stretch in Ph is critical to prevent nonproductive aggregation of both Drosophila and human Ph via their C-terminal sterile alpha motif (SAM) domains in vitro. Full Ph repressor activity in vivo requires both the SAM domain and O-GlcNAcylation of the S/T stretch. We demonstrate that Ph mutants lacking the S/T stretch reproduce the phenotype of ogt mutants, suggesting that the S/T stretch in Ph is the key Ogt substrate in Drosophila. We propose that O-GlcNAcylation is needed for Ph to form functional, ordered assemblies via its SAM domain” (doi:10.1016/j.devcel.2014.10.020)
-
Methylation
-
Lactylation
“Zhao et al.identified 26 histone lysine lactylation sites in HeLa
cells and 18 sites in mouse bone marrow-derived macrophages ... It
is important to note that variations in the identified histone
lactylation sites may arise from species-specific or spatiotemporal
dynamics of lactylation. Crucial questions remain unanswered,
including the identification of specific chromosomal regions
enriched with histone lactylation sites and the elucidation of
biological processes linked to these modifications. In-depth
investigations are warranted to unveil these aspects and fully
comprehend the significance of histone lactylation”.
”Lactic acid, traditionally considered as a metabolic waste product arising from glycolysis, has undergone a resurgence in scientific interest since the discovery of the Warburg effect in tumor cells. Numerous studies have proved that lactic acid could promote angiogenesis and impair the function of immune cells within tumor microenvironments. Nevertheless, the precise molecular mechanisms governing these biological functions remain inadequately understood. Recently, lactic acid has been found to induce a posttranslational modification, lactylation, that may offer insight into lactic acid's non-metabolic functions. Notably, the posttranslational modification of proteins by lactylation has emerged as a crucial mechanism by which lactate regulates cellular processes" (Hu, He, Li et al. 2025, doi:10.1186/s13148-024-01682-2).
-
General considerations
-
Some further general considerations
- A whole additional level of regulation is supplied by the enzymes that apply or remove these various chemical groups — for example, histone acetylases and deacetylases, demethylases, and so on. And these in turn are subject to post-translational modifications affecting their function. ...
- “Long noncoding RNAs have also been shown to be necessary for targeting histone-modifying activities. ... Histone methylation [can be] the end result of transcription of long noncoding RNAs and the subsequent nucleation and targeting of histone modifying complexes” (Zentner and Henikoff 2013).
- ...Then there are the acetyl and methyl groups (for example) that these enzymes apply to the histones. These groups are metabolites “whose availability and intracellular localization may dictate the efficacy and specificity of the enzymatic reaction”. That is, the epigenetic processes involving histone modifications are thus linked to metabolism. It’s possible that "distinct metabolites may localize to chromatin subdomains, favoring the clustering of relevant posttranslational modifications at specific genomic loci. The presence of metabolite ‘niches’ within specific chromatin subdomains has been proposed and is conceptually intriguing when placed in parallel with the idea of nuclear subcompartments and transcription ‘hubs’” (Sassone-Corsi 2013).
- “An interesting case has been reported of the combined effect of a histone variant (H2A.Z), a histone modification (H3K9Me), and a chromatin remodeling protein (HP1), all of which act to increase chromatin compaction”. This suggests the need to reckon with the “synergistic effects of histone variants” (Woodcock and Ghosh 2010, p. 8).
- It is “becoming clear that signalling events target proteins with histone tail-like sequences, dubbed ‘histone mimics’”. These mimics can undergo post-translational modifications just like the histone tails themselves, and they can attract some of the same proteins that “read” tail modifications and bind to the tails. “One possibility, therefore, is that histone mimics might allow a single signalling event to coordinate changes on chromatin by co‐modifying not only histones but also their regulators” (Badeaux and Shi 2013).
- The following offers, via a single detail, a hint of the complexity relating to the assembly of histones and their deposition onto DNA, and the significance of these processes for gene expression: “Nucleosome assembly in vivo requires assembly factors, such as histone chaperones, to bind to histones and mediate their deposition onto DNA. In yeast, the essential histone chaperone FACT functions in nucleosome assembly and H2A-H2B deposition during transcription elongation and DNA replication ... we report that the histone H2B repression (HBR) domain within the H2B N-terminal tail is important for histone deposition by FACT. Deletion of the HBR domain causes significant defects in histone occupancy in the yeast genome, particularly at HBR-repressed genes, and a pronounced increase in H2A-H2B dimers that remain bound to FACT in vivo. Moreover, the HBR domain is required for purified FACT to efficiently assemble recombinant nucleosomes in vitro. We propose that the interaction between the highly basic HBR domain and DNA plays an important role in stabilizing the nascent nucleosome during the process of histone H2A-H2B deposition by FACT” (Mao, Kyriss, Hodges et al. 2016, doi:10.1093/nar/gkw588).
-
Relation to DNA replication and development. Histone
modifications must be both maintained and changed across cell
generations during development of specialized cell lineages from
the undifferentiated zygote. It’s looking like an ever more
complex business: “By contrast to the single mechanism for copying
genetic information by semi-conservative replication, recent
studies suggest that copying of the epigenetic information is a lot
more complicated and varied. In some cases, such as the dilution
model, the histone modifications do indeed appear to be directly
inherited from the parental chromatin. In other instances, distinct
mechanisms exist to re-establish different histone marks after DNA
replication. In some cases, the histone-modifying enzyme is
recruited to the replication fork, while in other cases the
histone-modifying enzyme itself is maintained on the DNA through
DNA replication. In other cases, the histone modifications are
re-established in a much less immediate manner throughout the cell
cycle. Although not mutually exclusive, sequence-specific DNA
binding factors also presumably re-recruit histone modifiers to the
chromatin to reestablish histone modification patterns. Presumably
the mechanism that is used to inherit or re-establish each histone
post-translational modification depends on the immediacy and
accuracy required by the cell for the presence of that particular
epigenetic mark” (Budhavarapu, Chavez and Tyler 2013). In other
words, everything is highly context-specific.
- “Epigenetic modifications undergo drastic erasure and reestablishment after fertilization. This reprogramming is required for proper embryonic development and cell differentiation. In mammals, some histone modifications are not completely reprogrammed and play critical roles in later development. In contrast, in nonmammalian vertebrates, most histone modifications are thought to be more intensively erased and reestablished by the stage of zygotic genome activation (ZGA). However, histone modifications that escape reprogramming in nonmammalian vertebrates and their potential functional roles remain unknown. Here, we quantitatively and comprehensively analyzed histone modification dynamics during epigenetic reprogramming in Japanese killifish, medaka (Oryzias latipes) embryos. Our data revealed that H3K27ac, H3K27me3, and H3K9me3 escape complete reprogramming, whereas H3K4 methylation is completely erased during cleavage stage. Furthermore, we experimentally showed the functional roles of such retained modifications at early stages: (i) H3K27ac premarks promoters during the cleavage stage, and inhibition of histone acetyltransferases disrupts proper patterning of H3K4 and H3K27 methylation at CpG-dense promoters, but does not affect chromatin accessibility after ZGA;(ii) H3K9me3 is globally erased but specifically retained at telomeric regions, which is required for maintenance of genomic stability during the cleavage stage. These results expand the understanding of diversity and conservation of reprogramming in vertebrates, and unveil previously uncharacterized functions of histone modifications retained during epigenetic reprogramming” (Fukushima, Takeda and Nakamura 2023; doi:10.1101/gr.277577.122).
- Caveat. Like molecular biology as a whole, the study of histone modifications has been plagued by habitual attempts to make particular, well-defined “causes” out of particular modifications or combinations of them. (This is behind the search for a histone “code”.) It is becoming more and more evident that the various correlations between histone modifications and gene expression (a few of which are mentioned above) have no simple or absolute causal significance, but are part of a larger and more complex picture that must be elucidated in the various concrete situations that occur. Rando 2012 is useful for pointing out some of the puzzles in the current understanding of histone modifications.
-
Histone tail modifications
-
DNA methylation versus histone modifications
Researchers have compared DNA methylation to H3K27 tri-methylation in
different tissues and throughout human development. DNA methylation is
a more complex process than histone methylation, and, consistent with
that, it appears that DNA methylation is used to silence key
developmental genes later in development, when they need to be
repressed more or less permanently. H3K27me3, on the other hand, is
often used to repress genes that may need to be activated at multiple
times during development.
-
Core histones and their modifications
“Many core PTMs map to residues located on the lateral surface of the
histone octamer, close to the DNA, and they have the potential to
alter intranucleosomal histone-DNA interactions. ... Whereas
modifications in the histone tails might have limited structural impact
on the nucleosome itself and function as signals to recruit specific
binding proteins, PTMs in the lateral surface can have a direct
structural effect on nucleosome and chromatin dynamics, even in the
absence of specific binding proteins” (Tropberger and Schneider 2013).
“In contrast to those present in histone tails, modifications in the core
regions of the histones had remained largely uncharacterised until
recently, when some of these modifications began to be analysed in
detail. Overall, recent work has shown that histone core modifications
can not only directly regulate transcription, but also influence
processes such as DNA repair, replication, stemness, and changes in cell
state”. “Novel modifications, such as arginine methylation, are also
present [on the core histones] and can directly affect the compaction of
the DNA coating the nucleosome” (Lawrence, Daujat and Schneider 2016,
doi:10.1016/j.tig.2015.10.007).
“Controlled modulation of nucleosomal DNA accessibility via
post-translational modifications (PTM) is a critical component to many
cellular functions. Charge-altering PTMs in the globular histone
core—including acetylation, phosphorylation, crotonylation,
propionylation, butyrylation, formylation, and citrullination—can alter
the strong electrostatic interactions between the oppositely charged
nucleosomal DNA and the histone proteins and thus modulate accessibility
of the nucleosomal DNA, affecting processes that depend on access to the
genetic information, such as transcription”.
Based on a model: “The predicted effect of charge-altering PTMs on DNA accessibility can vary dramatically, from virtually none to a strong, region-dependent increase in accessibility of the nucleosomal DNA ... Proximity to the DNA is suggestive of the strength of the PTM effect, but there are many exceptions. For the vast majority of charge-altering PTMs, the predicted increase in the DNA accessibility should be large enough to result in a measurable modulation of transcription. However, a few possible PTMs, such as acetylation of H4K77, counterintuitively decrease the DNA accessibility, suggestive of the repressed chromatin ... For the majority of charge-altering PTMs, the effect on DNA accessibility is simply additive (noncooperative), but there are exceptions, e.g., simultaneous acetylation of H4K79 and H3K122, where the combined effect is amplified” (Fenley, Anandakrishnan, Kidane and Onufriev 2018, doi:10.1186/s13072-018-0181-5).
- Lysine acetylation of H3K122 near the dyad axis of the histone octamer has a direct affect in stimulating transcription. It presumably neutralizes the local positive charge on the histone surface, thereby loosening the DNA-protein binding there and making it easier for transcription-related factors to get access to the DNA. Acetylation of H3K122 “is specifically enriched at active transcription start sites as well as on [variant histone] H3.3- and H2A.Z-containing nucleosomes”. Those variant histones also are known to play a role in destabilizing nucleosomes and increasing access to DNA. In the particular type of cells studied, “H3K122ac is dynamically regulated at estrogen-regulated genes and marks enhancers that are actively engaged in transcriptional regulation” (Tropberger, Pott, Keller et al. 2013).
- “Besides H3K122, other lysines on the lateral surface of H3 — in particular, H3K56, H3K64, and H3K115 — can also be modified and might act synergistically and/or in different combinations, increasing the impact on nucleosome dynamics. Additionally, phosphorylation on the lateral surface could have a similar effect in reducing the DNA-binding affinity [with the histone]. ... As our work demonstrates, modifications on the lateral surface of the nucleosome are of central importance for chromatin biology, and we are just beginning to understand their mechanism of action and their role in the regulation of transcription” (Tropberger, Pott, Keller et al. 2013).
- Lysine 56 on the H3 histone (H3K56) is located at the entry-exit point of the enwrapped DNA. Its acetylation enables “breathing” of the DNA on the nucleosome core. (See Nucleosome wrapping and unwrapping below.) This facilitates access to that portion of the DNA by transcription factors and other regulatory elements. But it also, as Tessarz and Kouzarides (2014) point out, makes for a different, more loosely organized form of chromatin. The belief is that “H3K56 acetylation is one of the mechanisms used to keep nucleosome-free chromatin regions accessible at the higher order level”.
- Acetylation can also help to destabilize nucleosomes. In particular, acetylation of H4K91 “decreases the association of H2A-H2B dimers with chromatin and can lead to nucleosome instability” (Tessarz and Kouzarides 2014).
- The range of known histone core modifications with implications for gene expression looks set to expand in much the way that the number and variety of histone tail modifications have hugely expanded over the past decade or two. Phosphorylation of a threonine residue on histone H3 (H3T118) “enhances DNA accessibility on the nucleosome dyad, nucleosome mobility and nuceosome disassembly”. It may also “induce the formation of alternative nucleosome arrangements”. Methylation of lysine 79 on histone H3 (H3K79) “has been shown to correlate with active transcription in yeast and mammalian cells”. The contextual complexity of such modifications is illustrated by the further explanation that “The structure of chemically dimethylated H3K79 showed that this modification does not cause a major change in nucleosome structure, but a subtle reorientation of the region surrounding Lys79, which probably results in the loss of a single hydrogen bond to the L2 loop of H4. The modified residue becomes almost completely accessible to the solvent, which indicates that it might generate a docking site [for other factors] rather than cause larger structural rearrangements within the nucleosome core” (Tessarz and Kouzarides 2014).
- On another front, acetylation and/or methylation of certain core histone residues can affect interaction between the histones and histone chaperones, thereby affecting chaperone-mediated nucleosome assembly (Tessarz and Kouzarides 2014).
- “Here we report a new layer of regulation in transcriptional elongation that is conserved from yeast to mammals. This regulation is based on the phosphorylation of a highly conserved tyrosine residue, Tyr 57, in histone H2A and is mediated by the unsuspected tyrosine kinase activity of casein kinase 2 (CK2).” Both the H2A tyrosine phosphorylation and the activity of CK2 appear to play a role in regulating and coordinating the deubiquitination activity of the SAGA complex during transcription. “Together, these results identify a new component of regulation in transcriptional elongation based on CK2-dependent tyrosine phosphorylation of the globular domain of H2A” (doi:10.1038/nature13736)
- “Previously, we identified eight amino acids in histones H3 and H4 that are required for nucleosome occupancy over highly transcribed regions of the genome ... We [now] find that histone H3 K122, Q120, and R49 are required for Spt2, Spt6, and Spt16 [histone chaperone] occupancies at genomic locations where transcription rates are high, but not over regions of low transcription rates. Furthermore, substitution at one residue, K122, located on the dyad axis of the nucleosome, results in improper reassembly and disassembly of nucleosomes, likely accounting for the transcription rate-dependent regulation by these mutant histones ... These data support a mechanism for histone chaperone binding where these factors interact with histone proteins to promote their activities during transcription” (Hainer and Martens 2016, doi:10.1186/s13072-016-0066-4).
-
Examples of crosstalk between core and tail histone modifications:
“H3K79me3 is found on genomic regions that are also enriched in
H3K4me3, indicating that both marks co-localise on active chromatin.
Similarly, H3K79me2-enriched regions also have increased H3K4me3.
However, it is unclear which mark is deposited first. Furthermore,
H3K79 methylation depends on the deposition of H2BK123Ub. H3K79me2
also has a reciprocal relationship with some modifications, for
example H4K16ac. The mutation of H4K16 to mimic permanent acetylation
reduces H3K79me2 levels, whereas the removal of H3K79me2 by Dot1
mutation increases the levels of H4 acetylation. Therefore, H3K79me2/3
marks co-localise with some marks and anticorrelate with others”
“Repressive lateral surface modifications can also interplay with histone tail modifications. For example, H3K64me3 co-localises with H3K9me3 on many genomic regions and the deletion of Suv39h1/2, the enzymes that catalyse H3K9me3, also reduces H3K64me3 levels. H3K64me3 relies on H3K9me3 for its deposition. However, some repetitive elements maintain their H3K64me3 status in Suv39h1/2–/– cells, indicating that H3K64me3 is not entirely dependent on H3K9me3 for its maintenance” (Lawrence, Daujat and Schneider 2016, doi:10.1016/j.tig.2015.10.007).
- Continuing documentation here of the unfolding drama of histone core and tail modifications is probably impractical and needless. What will be necessary is to recognize the character, pattern, and functional (meaningful) “behavior” of the fluid, dynamic, intensely interwoven choreography of which these modifications and a great deal else that is recorded in these notes are a part.
- “We report monoallelic missense mutations affecting lysine 91 in the histone H4 core (H4K91) in three individuals with a syndrome of growth delay, microcephaly and intellectual disability. Expression of the histone H4 mutants in zebrafish embryos recapitulates the developmental anomalies seen in the patients. We show that the histone H4 alterations cause genomic instability, resulting in increased apoptosis and cell cycle progression anomalies during early development. Mechanistically, our findings indicate an important role for the ubiquitination of H4K91 in genomic stability during embryonic development” (Tessadori, Giltay, Hurst et al. 2017, doi:10.1038/ng.3956).
-
Histone variants
The ever-growing number of known histone variants has been revealing
that “the nucleosome is not a static entity but rather flexible and
dynamic”. For example, there is a shifting between a more closed and a
more open state, where the several histones comprising the nucleosome
core are more tightly or less tightly bound together. Histone variants
contribute to this dynamism, and have large effects on chromatin
structure, and thereby on many aspects of gene expression. It is
commonly said that the histone core of a nucleosome is wrapped by 147
base pairs of DNA, but this “must rather be viewed as a ‘snapshot’ of
one possible state”, with the actual number of base pairs varies
between 100 and 170 (Bönisch and Hake 2012).
“Histone variants are distinguished from canonical histones not only by
their amino acid sequences and physical properties but also by their
incorporation into chromatin outside of replication. This ability to
use different deposition modes makes them adaptable to respond to
environmental stimuli, which typically are not synchronous with
replication” (Talbert and Henikoff 2014, doi:10.1016/j.tcb.2014.07.006).
“Histone variants endow chromatin with unique properties and show a
specific genomic distribution that is regulated by specific deposition
and removal machineries. These variants — in particular, H2A.Z, macroH2A
and H3.3 — have important roles in early embryonic development, and they
regulate the lineage commitment of stem cells, as well as the converse
process of somatic cell reprogramming to pluripotency. Recent progress
has also shed light on how mutations, transcriptional deregulation and
changes in the deposition machineries of histone variants affect the
process of tumorigenesis. These alterations promote or even drive cancer
development through mechanisms that involve changes in epigenetic
plasticity, genomic stability and senescence, and by activating and
sustaining cancer-promoting gene expression programmes”
(Buschbeck and Hake 2017, doi:10.1038/nrm.2016.166).
“Histone variants are characterized by a distinct protein sequence and a
selection of designated chaperone systems and chromatin remodelling
complexes that regulate their localization in the genome. In addition,
histone variants can be enriched with specific post-translational
modifications, which in turn can provide a scaffold for recruitment of
variant-specific interacting proteins to chromatin. Thus, through these
properties, histone variants have the capacity to endow specific regions
of chromatin with unique character and function in a regulated manner”
(Martire and Banaszynski 2020, doi:10.1038/s41580-020-0262-8).
“Expression of short histone H2A (sH2A) variants is largely limited to
the testes, where they modulate splicing and nucleosome stability.
Through analysis of existing transcriptomic datasets, Chew et al. reveal
that sH2As are upregulated and splicing patterns are altered in a broad
range of cancers, particularly lymphomas but also endometrial, bladder
and cervical carcinomas. Thus, sH2As act as oncohistones when aberrantly
expressed in tissues other than the testes, possibly owing to their
ability to disrupt chromatin”
(Clyde 2021, doi:10.1038/s41576-021-00331-1).
Histone variants have so many diverse effects in different contexts that we look here at only a random sampling:
- Variant histones can destabilize nucleosomes. This may make the enwrapped DNA more accessible to transcription factors or other regulatory molecules, and also may make it easier for the nucleosome core particle to slide along the DNA.
- “Among core histones, the H2A family exhibits highest sequence divergence, resulting in the largest number of variants known”. They differ mostly in their “docking domain”, strategically placed at the DNA entry/exit site and implicated in interactions with [other parts of the nucleosome]. Moreover, the acidic patch, important for internucleosomal contacts and higher-order chromatin structure, is altered between different H2A variants. Consequently, H2A variant incorporation has the potential to strongly regulate DNA organization on several levels resulting in meaningful biological output” (Bönisch and Hake 2012).
- Histone variant MacroH2A is important in the inactivation of one of the X chromosomes in female mammals. But it evidently has wider functions also: “MacroH2A is displaced from chromatin after fertilization, suggesting that exclusion of macroH2A from chromatin is associated with a period of genome-wide reprogramming in pre-implantation development. Moreover, [it is] likely that histone H2A variants have a major role in determining chromatin plasticity and developmental potential. Importantly, macroH2A might have a similar role in restricting gene expression for preventing tumourigenesis” (Wutz 2011).
- “Our studies show that H2A.Z and H3.3 delineate the orientation of transcription at enhancers as observed at promoters. We also showed that enhancers with skewed histone variant patterns well [sic] facilitate enhancer activity. Collectively, our study indicates that histone variants are deposited at regulatory regions to assist gene regulation” (Won, Choi, LeRoy et al. 2015, doi:10.1186/s13072-015-0005-9).
- There is an “‘epigenetic peculiarity’ in olfactory neurons involving the expression of a histone H2b isoform (or variant) named H2be. This histone variant, which differs by only five amino acids from the canonical H2b protein, appears to be a gauge of the external olfactory sensory environment by being exclusively expressed from understimulated olfactory neurons, signaling the shortening of their life span” (Lomvardas and Maniatis 2016, doi:10.1101/cshperspect.a024208).
-
Histone H3 variants
“The deposition of the replicative H3 variant following DNA
replication is essential for the transmission of the epigenomic
information encoded in posttranscriptional modifications. Through this
process, replicative H3 maintains cell fate while, in contrast, the
replacement H3.3 variant opposes cell differentiation during early
embryogenesis. In later steps of development, H3.3 and specialized H3
variants are emerging as new, important regulators of terminal cell
differentiation, including neurons and gametes. The specific pathways
that regulate the dynamics of the deposition of H3.3 are paramount
during reprogramming events that drive zygotic activation and the
initiation of a new cycle of development”
(Loppin and Berger 2020, doi:10.1146/annurev-genet-022620-100039)
- Histone H3 variants have been proposed to function as a “bar code” affecting local functions. “H3.3 was generally regarded as an active histone mark in that its presence correlated with gene activation. However a few recent studies showed that H3.3 was also involved in heterochromatin formation in ES (embryonic stem) cells” (Li and Reinberg 2011).
- Histone H3.3 “plays a key role during gametogenesis, fertilization, and cellular differentiation. H3.3 is specifically enriched at the TSS being coupled with transcriptional initiation. In particular, it is found associated with the high CpG/broad class of promoter, analogously to H2A.Z. It is also located in the gene body of active genes, where its abundance is directly proportional to transcriptional activity, as well as at CTCF and transcription factor binding sites located at enhancers ... H3.3 also localizes to sites of DNA damage to facilitate reactivation of transcription once repair is complete. Conversely, H3.3 is also found at repressed promoters, is required for heterochromatin formation in the mouse embryo, and plays an important role at telomeres in mouse ES cells. Its ability to reprogram chromatin is underpinned by the timing and its sites of incorporation. Mechanistically, H3.3 can modulate chromatin structure directly and/or indirectly depending upon its modification state and its ability to antagonize histone H1 incorporation” (Soboleva, Nekrasov, Ryan and Tremethick 2014).
- In a study of embryonic stem cells (ESCs): “H3.3 is found decorated with various histone modifications that regulate transcription and maintain chromatin integrity. We find greatly varying H3.3 dissociation rates across various histone modification domains: high dissociation rates at active histone marks and low dissociation rates at heterochromatic marks. Well-defined zones of high H3.3-nucleosome turnover were detected at binding sites of ESC-specific pluripotency factors and chromatin remodelers, suggesting an important role for H3.3 in facilitating protein binding. Among transcription factor binding sites we detected higher H3.3 turnover at distal cis-acting sites compared to proximal genic transcription factor binding sites ... The presence of high H3.3 turnover at RNA Pol II binding sites at extragenic regions as well as at transcription start and end sites of genes, suggests a specific role for H3.3 in transcriptional initiation and termination. On the other hand, the presence of well-defined zones of high H3.3 dissociation at transcription factor and chromatin remodeler binding sites point to a broader role in facilitating accessibility” (Ha, Kraushaar and Zhao 2014, doi:10.1186/1756-8935-7-38).
- “The composition and structure of centromeric nucleosomes, which contain the histone H3 variant CENP-A, is intensely debated. Two independent studies in this issue [of Cell], in yeast and human cells, now suggest that CENP-A nucleosomes adopt different structures depending on the stage of the cell cycle” (Westhorpe and Straight 2012).
- The protein and histone chaperone, DAXX, together with other factors, deposits the histone variant H3.3 into telomeric and pericentromeric repeats. The latter are key to the formation of “spatially discrete, compact, constitutive heterochromatic structures called chromocentres [which] serve as integral, functionally important components of nuclear organization”. DAXX turns out to be a “major regulator of subnuclear organization through the maintenance of the global heterochromatin structural landscape”. “We show, for the first time, that the loss of a histone chaperone can have severe consequences for global nuclear organization and chromatin sensitivity” (Rapkin, Ahmed, Dulev et al. 2015, doi:10.1186/s13072-015-0036-2).
- “Enriched integration of histone H3.3, the ancestral histone H3 variant, is a general feature of dynamically regulated chromatin and transcription4,5,6,7. However, how chromatin is regulated at induced genes, and what features of H3.3 might enable rapid and high-level transcription, are unknown. The amino terminus of H3.3 contains a unique serine residue (Ser31) that is absent in ‘canonical’ H3.1 and H3.2. Here we show that this residue, H3.3S31, is phosphorylated (H3.3S31ph) in a stimulation-dependent manner along rapidly induced genes in mouse macrophages. This selective mark of stimulation-responsive genes directly engages the histone methyltransferase SETD2, a component of the active transcription machinery, and ‘ejects’ the elongation corepressor ZMYND118,9. We propose that features of H3.3 at stimulation-induced genes, including H3.3S31ph, provide preferential access to the transcription apparatus. Our results indicate dedicated mechanisms that enable rapid transcription involving the histone variant H3.3, its phosphorylation, and both the recruitment and the ejection of chromatin regulators” (Armache, Yang, Martínez de Paz et al. 2020, doi:10.1038/s41586-020-2533-0).
- “Although the promyelocytic leukemia (PML) protein is renowned for regulating a wide range of cellular processes and as an essential component of PML nuclear bodies (PML-NBs), the mechanisms through which it exerts its broad physiological impact are far from fully elucidated. Here, we review recent studies supporting an emerging view that PML’s pleiotropic effects derive, at least partially, from its role in regulating histone H3.3 chromatin assembly, a critical epigenetic mechanism. These studies suggest that PML maintains heterochromatin organization by restraining H3.3 incorporation. Examination of PML’s contribution to H3.3 chromatin assembly in the context of the cell cycle and PML-NB assembly suggests that PML represses heterochromatic H3.3 deposition during S phase and that transcription and SUMOylation regulate PML’s recruitment to heterochromatin” (Delbarre and Janicki 2021, doi:10.1002/bies.202100038).
- “H3.3 is a replication-independent H3 histone variant in mammalian systems that is enriched at both H3K4me3- and H3K27me3-marked bivalent genes as well as H3K9me3-marked endogenous retroviral repeats. Here we show that H3.3, but not its chaperone Hira, prevents premature HSC [haematopoietic stem cell] exhaustion and differentiation into granulocyte-macrophage progenitors. H3.3-null HSPCs [haematopoietic stem progenitor cells] display reduced expression of stemness and lineage-specific genes with a predominant gain of H3K27me3 marks at their promoter regions. Concomitantly, loss of H3.3 leads to a reduction of H3K9me3 marks at endogenous retroviral repeats, opening up binding sites for the interferon regulatory factor family of transcription factors, allowing the survival of rare, persisting H3.3-null HSCs. We propose a model whereby H3.3 maintains adult HSC stemness by safeguarding the delicate interplay between H3K27me3 and H3K9me3 marks, enforcing chromatin adaptability” (Guo, Liu, Geng et al. 2022, doi:10.1038/s41556-021-00795-7).
-
Histone variant H2A.Z
One more example of a histone variant:- H2A.Z can also play a role in both repression and activation of gene expression. For example, mono-ubiquitylation of H2A.Z is linked to transcriptional silencing, while deubiquitylation promotes gene activation (Draker, Sarcinella and Cheung 2011).
- Variant histone H2A.Z in the nucleosome immediately downstream of the transcription start site of active genes serves to “mark” the gene (which is temporarily inactivated during mitosis) for reactivation following mitosis (Kelly, Miranda, Liang et al. 2010).
- “H2A.Z promotes formation of the higher-order chromatin fiber in a manner dependent upon just two amino acid residues [of the histone], which subtly extend the acidic patch of H2A.Z compared to that of H2A, and cooperate with heterochromatin protein HP1α to establish or maintain a specialized conformation at constitutive heterochromatic [and generally gene-repressive] domains” (Li and Reinberg 2011).
- “Histone variant H2A.Z antagonizes DNA methylation along the whole genome in plants and animals” (Li and Reinberg 2011).
- Summarizing histone variant H2A.Z: “H2A.Z has multiple roles in regulating transcription and the ultimate outcome may depend upon whether its primary function is based at the promoter, the TSS, or in the gene body. Indeed, based on these different locations, H2A.Z might potentially have opposing and competing functions even on the same gene. Further regulation could be achieved depending upon whether the H2A.Z-containing nucleosome is heterotypic or homotypic (or cycling between these two states). Finally, adding to this complexity, H2A.Z can be post-translationally modified. H2A.Z acetylation and ubiquitylation have been shown to be associated with gene activation and repression, respectively” (Soboleva, Nekrasov, Ryan and Tremethick 2014).
- “The histone variant H2A.Z has been extensively studied to understand its manifold DNA-based functions. In the past years, researchers identified its specific binding partners, the ‘H2A.Z interactome’, that convey H2A.Z-dependent chromatin changes. Here, we summarize the latest findings regarding vertebrate H2A.Z-associated factors and focus on their roles in gene activation and repression, cell cycle regulation, (neuro)development, and tumorigenesis. Additionally, we demonstrate how protein–protein interactions and post-translational histone modifications can fine-tune the complex interplay of H2A.Z-regulated gene expression. Last, we review the most recent results on interactors of the two isoforms H2A.Z.1 and H2A.Z.2.1, which differ in only three amino acids, and focus on cancer-associated mutations of H2A and H2A.Z, which reveal fascinating insights into the functional importance of such minuscule changes”. “H2A.Z and its interactors ... are emerging as novel regulators in early development and neurodevelopment, associate with diseases as well as cancer development, and offer new therapeutic potential” (Kreienbaum, Paasche and Hake 2022, doi:10.1016/j.tibs.2022.04.014).
-
The complexity of the role of H2A.Z in gene regulation. “The androgen receptor and estrogen receptor systems represent good examples to explain the function of H2A.Z in gene transcription. In the androgen receptor system, the PSA gene can be considered as the prototype of this pathway: In the absence of androgen (OFF [repressed] state), H2A.Z is loaded by the SRCAP and/or p400/Tip60 complexes. In this repressed configuration, H2A.Z is monoubiquitinated at both enhancers and promoters potentially by RING1B. Upon androgen stimulation (ON), H2A.Z is deubiquitinated by USP10 and its occupancy decreases. Of note, H2A.Z acetylation correlates with androgen receptor induction and similarly, the occupancy of the p400/Tip60 complex increases upon androgen receptor induction. The recruitment of the p400/Tip60 complex is mediated by its MRG15 subunit which recognizes H3K4 methylation states while SRCAP has been shown to interact with androgen receptor. In the case of the estrogen signaling cascade, we focus on the case of the TFF1 locus: In the OFF state, forkhead box protein A1 (FoxA1) binds to a distal enhancer (FoxA1-binding site) of the TFF1 locus where it recruits the p400/Tip60 complex supporting H2A.Z loading. Lack of H2A.Z at the TFF1 promoter, leads to a poorly defined nucleosome occupancy in the repressed/poised state (OFF). Upon activation of the pathway, the p400/Tip60 complex is recruited at the TFF1 promoter by estrogen receptor α which binds to its cognate sequences. At the TFF1 promoter, the p400/Tip60 complex loads H2A.Z leading to a better-defined nucleosome positioning. At the same time, H2A.Z occupancy decreases at the FoxA1-bound distal enhancer.
“From the above, some general rules for H2A.Z in gene regulation can be postulated: At genes that are poised/repressed (OFF), repressive marks of H2A.Z are found and as consequence its loss of function leads to upregulation. At genes that are active, activating PTMs of H2A.Z, such as H2A.Z acetylation, are found and as consequence H2A.Z loss of function leads to downregulation.
“In a repressed (OFF) or poised state, the H2A.Z deposition machinery is recruited by transcription factors and/or histone modifications to chromatin. This recruitment can be transient but still allows an exchange of H2A with H2A.Z. In the OFF state, H2A.Z is deacetylated by the deacetylation machinery and ubiquitinated on its C-terminus by RING1B. Upon gene activation (ON), additional TFs and/or histone modifications lead to the recruitment of the loading/acetylation/deubiquitination machinery. This triggers H2A.Z acetylation and deubiquitination, finally leading to transcriptional activation” (Giaimo, Ferrante, Herchenröther, et al. 2019, doi:10.1186/s13072-019-0274-9)
All this barely hints at the complexity of histone regulation overall.
-
Seasonal response of histone variants
-
“Many organisms undergo profound changes in gene expression
with the seasons. In the common carp, Cyprinus carpio, a
notable seasonal morphological change in the nucleolus of
hepatocytes correlates with changes in rRNA transcription,
which is highest in the summer. During winter, downregulation
of rRNA is accompanied by hypermethylation of the ribosomal
cistron. H2A.Z levels are increased overall in these cells
during winter, but at the ribosomal cistron H2A.Z is increased
during summer. Ubiquitylation of H2A.Z, which is usually
associated with gene silencing, was also enriched at the
ribosomal cistron during summer. This suggests multiple layers
of seasonal regulation” (Talbert and Henikoff 2014,
doi:10.1016/j.tcb.2014.07.006).
“Similarly to other vertebrates, carp has two macroH2A genes. MacroH2A.1 is enriched at the ribosomal cistron and at the promoter of the L41 ribosomal protein gene during winter. Enrichment of macroH2A.1 at these sites colocalizes with enrichment for H3K27 methylation, a mark of repressed chromatin. Consistent with this, macroH2A.1 represses rDNA transcription in human cells” (Talbert and Henikoff 2014, doi:10.1016/j.tcb.2014.07.006).
- “In summer the ribosomal cistron and L41 are instead enriched for macroH2A.2 and H3K4me3, a mark of active chromatin, consistent with the increased transcription of both loci. By contrast, no seasonal change is seen in macroH2A.1 or macroH2A.2 at the prolactin gene promoter. Although the roles of macroH2A.1 and macroH2A.2 are not well understood, these observations suggest that they may have opposing or complementary roles in gene expression” (Talbert and Henikoff 2014, doi:10.1016/j.tcb.2014.07.006).
-
“Many organisms undergo profound changes in gene expression
with the seasons. In the common carp, Cyprinus carpio, a
notable seasonal morphological change in the nucleolus of
hepatocytes correlates with changes in rRNA transcription,
which is highest in the summer. During winter, downregulation
of rRNA is accompanied by hypermethylation of the ribosomal
cistron. H2A.Z levels are increased overall in these cells
during winter, but at the ribosomal cistron H2A.Z is increased
during summer. Ubiquitylation of H2A.Z, which is usually
associated with gene silencing, was also enriched at the
ribosomal cistron during summer. This suggests multiple layers
of seasonal regulation” (Talbert and Henikoff 2014,
doi:10.1016/j.tcb.2014.07.006).
-
Histone variants at the transcription start site
- “A long-held view has been that the TSS [transcription start site] is positioned within a ‘naked’ DNA region. However, new data show that, from simple to higher eukaryotes, the TSS is not histone-free but is associated with an unstable and nuclease-sensitive nucleosome. Further, this specialized nucleosome is marked by the incorporation of specific histone variants in higher eukaryotes (Soboleva, Nekrasov, Ryan and Tremethick 2014).
- “The function of this unstable nucleosome [at transcription start sites] remains to be determined, but although it might not impede binding of Pol II enzyme it might concomitantly serve as a ‘placeholder’ to keep the TSS in an accessible state. The presence of an unstable nucleosome at the TSS may also provide a mechanism that can regulate the level of transcription depending upon the rate or level of histone variant exchange” (Soboleva, Nekrasov, Ryan and Tremethick 2014).
- "We identify the mouse (Mus musculus) H2A histone variant H2A.Lap1 as a previously undescribed component of the TSS [transcription start site] of active genes expressed during specific stages of spermatogenesis. This unique chromatin landscape also includes a second histone variant, H2A.Z. In the later stages of round spermatid development, H2A.Lap1 dynamically loads onto the inactive X chromosome, enabling the transcriptional activation of previously repressed genes. Mechanistically, we show that H2A.Lap1 imparts unique unfolding properties to chromatin. We therefore propose that H2A.Lap1 coordinately regulates gene expression by directly opening the chromatin structure of the TSS at genes regulated during spermatogenesis” (Soboleva, Nekrasov, Pahwa et al. 2012).
-
Acidic patch of the nucleosome core particle
-
The histone core particle has a small acidic (negatively
charged) patch formed by six acidic residues from H2A and one
from H2B. “Neutralization of only three acidic amino acid
residues within this patch was sufficient to inhibit the
intra-nucleosome–nucleosome interactions necessary for the
compaction of chromatin. The acidic patch on a nucleosome
mediates the compaction of chromatin by interacting with the
histone H4 N-terminal tail originating from a neighboring
nucleosome. Therefore, remarkably, subtle charge and/or
stereochemical changes to the surface of the nucleosome in this
region can have a profound effect on the protein-protein
interactions that govern chromatin compaction.
“The eukaryotic cell has devised ways to alter the acidic patch to regulate chromatin structure and function. The replacement of both copies of canonical H2A with H2A.Z promotes chromatin compaction, and this ability is dependent upon H2A.Z creating an acidic patch that is slightly more acidic than H2A. By contrast, incorporation of H2A.Bbd or H2A.Lap1 into nucleosome arrays completely inhibits array folding, which is due to H2A.Bbd/H2A.Lap1 generating an acidic patch that is less acidic than H2A” (Soboleva, Nekrasov, Ryan and Tremethick 2014).
-
The histone core particle has a small acidic (negatively
charged) patch formed by six acidic residues from H2A and one
from H2B. “Neutralization of only three acidic amino acid
residues within this patch was sufficient to inhibit the
intra-nucleosome–nucleosome interactions necessary for the
compaction of chromatin. The acidic patch on a nucleosome
mediates the compaction of chromatin by interacting with the
histone H4 N-terminal tail originating from a neighboring
nucleosome. Therefore, remarkably, subtle charge and/or
stereochemical changes to the surface of the nucleosome in this
region can have a profound effect on the protein-protein
interactions that govern chromatin compaction.
-
Histone turnover
In yeast, heterochromatin and euchromatin chromosome domains have been
found to be related to histone turnover, with euchromatin (favorable to
gene expression) associated with more rapid histone turnover (Aygün,
Mehta and Grewal 2013).
“The association of histones with specific chaperone complexes is
important for their folding, oligomerization, post-translational
modification, nuclear import, stability, assembly and genomic
localization. In this way, the chaperoning of soluble histones is a key
determinant of histone availability and fate, which affects all
chromosomal processes, including gene expression, chromosome segregation
and genome replication and repair ... Chaperones cooperate in the
histone chaperone network and via co-chaperone complexes to match histone
supply with demand, thereby promoting proper nucleosome assembly and
maintaining epigenetic information by recycling modified histones evicted
from chromatin” (Hammond, Strømme, Huang et al., doi:10.1038/nrm.2016.159)
- In a typical sort of crosstalk, histone tail acetylation correlates with higher turnover rates, while deacetylation correlates with lower turnover rates and heterochromatin formation (Aygün, Mehta and Grewal 2013). There are, of course, various other factors that play roles relevant to acetylation.
-
Nucleosome wrapping and unwrapping
The mutual embrace of DNA and the core histones of a nucleosome is
rhythmically relaxed at certain positions — especially at the points
where DNA enters and exits the histone complex. This leads to a
partial unwrapping of the DNA from the histones and allows readier
access of DNA-binding factors affecting transcription. “The
equilibrium between the fully wrapped and partially wrapped nucleosome
states is termed nucleosome site exposure, and conversion into a
partially unwrapped nucleosome occurs many times per second” (North,
Shimko, Javaid et al. 2012). This wrapping and unwrapping is
sometimes referred to as “DNA breathing” (as in the paragraph
immediately following), a term that is confusing because one also hears
of the “breathing” of
Hoogsteen base pairs,
as well as a “DNA breathing”
whereby the strands of double-stranded DNA temporarily separate and
reunite at particular loci. The wrapping and unwrapping of DNA in
nucleosomes also should not be confused with
chromatin breathing — although the two
processes may be related.
“The apparent homogeneity and stability of nucleosomes has led to their
depiction as beads, balls, and other simplifications that imply a largely
static histone structural surface on which DNA wraps and unwraps. [New
researches] enrich our understanding of nucleosome behavior with direct
evidence that the histone octamer must itself flex to undergo chromatin
remodeling, a common step in many genome transactions”
(Flaus and Owen-Hughes 2016, doi:10.1126/science.aam5403).
- “The frequency of DNA breathing (i.e. spontaneous, localized release of DNA contact with histones) of the first ~20 base pairs occurs once every 250 ms, but the frequency of DNA breathing ~40 base pairs into the nucleosome progressively and rapidly decreases to once every 10 min and even longer closer to the nucleosome dyad” (Petesch and Lis 2012).
- This process, unsurprisingly, is related to other regulatory features. For example: “Acetylation of H3K56 increases DNA breathing of the nucleosome ~40 base pairs away from the dyad by sevenfold, allowing DNA that is less tightly wound to gain easier access to proteins such as Pol II” (Petesch and Lis 2012). The DNA sequence within the entry and exit regions of the nucleosome also affects the unwrapping rate (North, Shimko, Javaid et al. 2012).
- In a yeast (Saccharomyces cerevisiae), approximately 30% of transcription factor binding sites reside in the nucleosome entry-exit region, so that modulation of the unwrapping rate looks like a factor in the regulation of gene expression (North, Shimko, Javaid et al. 2012).
- “we observed that the nucleosome can unwrap asymmetrically and directionally under force. The relative DNA flexibility of the inner quarters of nucleosomal DNA controls the unwrapping direction such that the nucleosome unwraps from the stiffer side ... The opening of one end helps to stabilize the other end, providing a mechanism to amplify even small differences in flexibility to a large asymmetry in nucleosome stability”. This has implications for gene regulation in various regards, one of which has to do with the choice between antisense and sense transcription: “Our results [suggest] the possibility that nature selects for lower flexibility DNA sequences within the first half of nucleosomes in the direction of transcription. In this scenario, RNA polymerase would have greater initial access to the DNA template if it enters the nucleosomal DNA from the ‘weak’ side and would only pause when it reaches the nucleo- somal dyad” (Ngo, Zhang, Zhou et al. 2015, doi:10.1016/j.cell.2015.02.001).
- Given that one end of the DNA can be more strongly bound to the nucleosome core particle than the other (see previous item), “a transient unwrapping of the strong side is often observed, and this is followed by rewrapping of the strong side and major unwrapping of the weak side in a coordinated fashion” (Ngo, Zhang, Zhou et al. 2015, doi:10.1016/j.cell.2015.02.001).
- “Previous biochemical studies have demonstrated that in the presence of adenosine triphosphate (ATP) the human RAD51 (HsRAD51) recombinase can form a nucleoprotein filament (NPF) on double-stranded DNA (dsDNA) that is capable of unwrapping the nucleosomal DNA from the histone octamer ... We show that oligomerization of HsRAD51 leads to stepwise, but stochastic unwrapping of the DNA from the histone octamer in the presence of ATP. The highly reversible dynamics observed in single-molecule trajectories suggests an antagonistic mechanism between HsRAD51 binding and rewrapping of the DNA around the histone octamer. These stochastic dynamics were independent of the nucleosomal DNA sequence or the asymmetry created by the presence of a linker DNA. We also observed sliding and rotational oscillations of the histone octamer with respect to the nucleosomal DNA. These studies underline the dynamic nature of even tightly associated protein–DNA complexes such as nucleosomes” (Senavirathne, Mahto, Hanne et al. 2017, doi:10.1093/nar/gkw920).
-
Nucleosome structural plasticity, asymmetry, and conformational shifts
Nucleosomes display varying degrees of stability, apparently owing to
a variety of biochemical factors, which may include histone core
particles and variants, chromatin structure, and rigidity of the
associated DNA.
New research represent a “general change in perspective from the
prevailing view that DNA deforms itself to slide across the rigid histone
surface, paralleling the static lock and key model of enzymes and
substrates. Instead, the requirement for flexibility within the histone
octamer suggests an equivalent of an induced-fit mechanism where
histone-histone and histone-DNA interactions deform to achieve a
transition state for nucleosome repositioning. Because the nucleosome
responds in different ways to the action of different enzymes, the
histone octamer substrate clearly plays a more active role in remodeling
than a simple bead on a chromatin string”
(Flaus and Owen-Hughes 2016, doi:10.1126/science.aam5403).
- In yeast a rather large class of “fragile nucleosomes” has been uncovered. “The fact that the fragile nucleosomes are highly enriched at various functional regions in the genome including the promoters of protein-coding genes, the tRNA genes, and replication origins, as well as LTRs [long terminal repeats], strongly suggests that nucleosome fragility is broadly implicated in many important chromatin-related processes”. This reveals “a new level of complexity in nucleosome organization” (Xi, Yao, Chen et al. 2011).
- In the case of environmental-stress-response genes, it has been proposed that “nucleosome fragility poises genes for swift up-regulation in response to the environmental changes” (Xi, Yao, Chen et al. 2011).
- The high mobility group protein HMGB1 serves to “relax” the nucleosome, in part by interacting electrically with the minor groove of the enwrapping DNA and increasing the flexure of the DNA. This has been shown to play a role in dramatically increasing the binding of the ER (estrogen receptor) transcription factor to nucleosomal DNA. HMGB1 increases binding of many other transcription factors as well (Joshi, Sarpong, Peterson and Scovell 2012).
- Regarding different nucleosomal conformations: evidence points to “a dynamic equilibrium of multiple populations of conformational isomers on a nucleosome energy landscape. This paradigm suggests that there is a statistical ensemble of nucleosome conformers in equilibrium, of which the population of states and the energy barriers between them is sensitive to the immediate microenvironment and to interactions from binding factors, such as HMGB1” (Joshi, Sarpong, Peterson and Scovell 2012).
- But there are many other possibilities for nucleosome forms. “Influences such as DNA methylation, posttranslational modifications of the core histone proteins, histone variants, SIN mutations and the level of chromatin compaction may each contribute to a multitude of additional energy states within the chromatin network. All these factors can potentially alter intra- and internucleosomal forces and establish a different or more extended ensemble of nucleosome conformational states, and therefore further fine-tune the functional activities. This is consistent with the notion of a heterogeneous population of nucleosomes within chromatin, all in a dynamic state and able to respond to continuous changes from environmental cues” (Joshi, Sarpong, Peterson and Scovell 2012).
- Strong evidence has been presented that nucleosomal particles occur not only as octamers, but also as “hexasomes” and half-nucleosomes (Rhee, Bataille, Zhang and Pugh 2014; McKay and Lieb 2014).
- The same researchers have demonstrated that the face of nucleosomes approached by the transcribing enzyme shows asymmetric patterns of histone variants and modifications compared to the face distal to the transcribing enzyme.
Linker histones are distinct from the histones making up the nucleosomal
core particle. A single such histone is capable of “tying together” the
DNA where that DNA enters and exits the core particle, thereby locking it
to the core histones. Alternatively, linker histones can loosen their hold
and facilitate an open chromatin environment.
“Histone H1 binding to chromatin has been shown to be dynamic in nature,
with specific H1 variants divergent in their binding affinity for
chromatin. It is thought that a high percentage of the total nuclear H1 is
bound to nucleosomes at any given time; however, these interactions are
individually transient. ... in vivo dynamics of histone H1.1 occur
through soluble intermediates, giving rise to a rapid ‘stop-and-go’
movement of H1.1 in the nucleus between random binding sites. Others have
further demonstrated that the transient binding of H1 variants with
nucleosomes is affected by the structure of the H1 variant,
post-translational modifications present on H1 and competition for
chromatin binding by other nuclear factors” (Harshman, Young, Parthun and
Freitas 2013).
[In mice:] “We show that the local density of H1 controls the balance of
repressive and active chromatin domains by promoting genomic compaction.
[A study of T cells] reveals that H1-mediated chromatin compaction occurs
primarily in regions of the genome containing higher than average levels of
H1 ... Reduction of H1 stoichiometry leads to decreased H3K27 methylation,
increased H3K36 methylation, B-to-A-compartment shifting [that is,
heterochromatin to euchromatin shifting] and an increase in interaction
frequency between compartments. In vitro, H1 promotes PRC2-mediated H3K27
methylation and inhibits NSD2-mediated H3K36 methylation. Mechanistically,
H1 mediates these opposite effects by promoting physical compaction of the
chromatin substrate. Our results establish H1 as a critical regulator of
gene silencing through localized control of chromatin compaction, 3D genome
organization and the epigenetic landscape”
(Willcockson, Healton, Weiss 2021, doi:10.1038/s41586-020-3032-z).
- By preventing the unwrapping of nucleosomal DNA (see “Nucleosome wrapping and unwrapping” above), and also by preventing rotation of the DNA double helix on the nucleosomal core particle, linker histones reduce access to the DNA by transcription factors and other regulatory complexes.
- The locking of the entering and exiting DNA together by linker histones can also aid in the formation of regularly spaced nucleosomes and therefore in the compaction of chromatin, which tends to repress the expression of genes within the compacted region.
- “Histone H1-bound nucleosomes can limit access of chromatin remodeling complexes” such as SWI/SNF. However, the data suggest that “specific remodeling complexes can access key nucleosomal elements without the removal of the linker histones” (Harshman, Young, Parthun and Freitas 2013).
- “Structural variation among histone H1 variants confers distinct modes of chromatin binding that are important for differential regulation of chromatin condensation, gene expression and other processes. Changes in the expression and genomic distributions of H1 variants during cell differentiation appear to contribute to phenotypic differences between cell types, but few details are known about the roles of individual H1 variants and the significance of their disparate capacities for phosphorylation ... Our data provide strong evidence that H1 variant interphase phosphorylation is dynamically regulated in a site-specific and gene-specific fashion during pluripotent cell differentiation, and that enrichment of pS187-H1.4 [phosphorylation at serine 187 of histone H1.4] at genes is positively related to their transcription. H1.4-S187 is likely to be a direct target of CDK9 during interphase, suggesting the possibility that this particular phosphorylation may contribute to the release of paused RNA pol II. In contrast, the other H1 variant phosphorylations we investigated appear to be mediated by distinct kinases and further analyses are needed to determine their functional significance” (Liao and Mizzen 2017, doi:10.1186/s13072-017-0135-3).
- “Linker histones (H1) bind to nucleosomes via electrostatic interactions and ... this binding can occur in either on-dyad or off-dyad mode. These different binding modes can lead to differential folding of nucleosome arrays, with different levels of compaction ... In addition to the regulation of chromatin structure, H1 is intimately involved in the control of multiple chromatin metabolism processes, such as DNA replication and repair, as well as modulation of the epigenetic landscape of the genome ... The occupancy of H1 in chromosomes is not uniform. The dynamic, locus-specific, activity- and cell cycle-dependent distribution of H1 has essential implications for its biological activities ... At the molecular level, H1 acts in a variety of distinct, biochemically separable mechanisms, including chromatin fibre compaction and limiting DNA accessibility to DNA-binding proteins, as well as tethering or specific inhibition of nuclear enzymes” (Fyodorov, Zhou, Skoultchi and Bai 2018, doi:10.1038/nrm.2017.94).
-
Linker histone variants and modifications
- “Mammals express up to 11 different H1 [linker] histone variants” which undergo many modifications, the significance of which is “largely unknown” (Weiss et al. 2010).
- Different linker histone modifications — specifically, methylations of distinct histone variants — have been demonstrated to have different effects, for example, on processes related to heterochromatin formation (Weiss et al. 2010).
- Acetylation of the linker histone can have an activating effect upon transcription: “H1.4K34 acetylation (H1.4K34ac) ... is preferentially enriched at promoters of active genes, where it stimulates transcription by increasing H1 mobility and recruiting a general transcription factor. H1.4K34ac is dynamic during spermatogenesis and marks undifferentiated cells such as induced pluripotent stem (iPS) cells and testicular germ cell tumors” (Kamieniarz, Izzo, Dundr et al. 2012).
- “Phosphorylation of histone H1 has many distinct functions, leading to both chromatin condensation and decondensation dependent on the site of phosphorylation and cell cycle context”. There seem to be two broad phases: “First, an interphase (G0–S phase) partial phosphorylation that allows for chromatin relaxation and facilitates transcriptional activation. Second, a maximal phosphorylation during mitosis (M phase) allows for chromatin condensation and separation of chromosomes into daughter cells. The partial phosphorylation observed in interphase has been shown to induce structural changes in the [C terminal domain tail] of H1, which in turn leads to a decreased affinity of histone H1 for DNA” (Harshman, Young, Parthun and Freitas 2013).
- “Phosphorylation of histone H1 has been shown to disrupt the interaction between itself and heterochromatin protein 1α, leading to chromatin condensation”.
- “While methylation and acetylation are the best-characterized histone post-translational modifications, citrullination by the protein arginine deiminases (PADs) represents another important player in this process. In addition to fine tuning chromatin structure at specific loci, histone citrullination can also promote rapid global chromatin decondensation during the formation of extracellular traps (ETs) in immune cells. Recent studies now show that PAD4-mediated citrullination of histone H1 at promoter elements can also promote localized chromatin decondensation in stem cells, thus regulating the pluripotent state. These observations suggest that PAD-mediated histone deimination profoundly affects chromatin structure, possibly above and beyond that of other post-translational modifications” (Slade, Horibata, Coonrod and Thompson 2014).
- Experiments with mice “revealed the essential function of [variant H1 histone] H1T2 for the DNA condensation and histone-to-protamine replacement in spermiogenesis ... Linker histone H1T2 possesses unique domain architecture which can account for the specific functions associated with chromatin remodeling events facilitating the initiation of histone-to-transition-proteins/protamine transition in the polar apical spermatid genome. Our results directly establish the unique function of H1T2 in nuclear shaping associated with spermiogenesis by mediating the interaction between chromatin and nucleo-skeleton, positioning the epigenetically specialized chromatin domains involved in transcription coupled histone replacement initiation towards the apical pole of round/elongating spermatids.” Analyses “revealed the open chromatin architecture of H1T2-occupied chromatin encompassing the H4 acetylation and other histone PTMs characteristic of transcriptionally active chromatin” (Shalini, Bhaduri, Ravikkumar et al. 2021, doi:10.1186/s13072-020-00376-2)
-
Linker histones and integrated regulation
- An example (from Vicent, Nacht, Font-Mateu, Castellano et al. 2011): “Within the first minute of progesterone action, a complex cooperation between different enzymes acting on chromatin mediates histone H1 displacement as a requisite for gene induction and cell proliferation": (1) the activated progesterone receptor recruits chromatin remodeling complexes to hormone target genes. (2) Trimethylation of histone H3 at Lys 4 by one of those complexes, enhanced by hormone-induced displacement of the H3K4 demethylase KDM5B, stabilizes one of the remodeling complexes, which then (3) facilitates the progesterone receptor-mediated recruitment of Cdk2/CyclinA, which in turn (4) mediates histone H1 displacement. This displacement “is required for hormone induction of most hormone target genes”.
“Much of the transcriptionally inactive portion of the mammalian genome is
located within condensed heterochromatin, predominantly at the nuclear
periphery. Around one third of the genome forms kilobase-to-megabase-sized
domains that are tethered to the nuclear lamina (lamina-associated domains;
LADs). These domains are enriched in heterochromatic histone modifications
and comprise gene-poor regions of low expression”
(Sexton 2019, doi:10.1016/j.tcb.2019.06.001).
Regarding a study by the van Steensel group: “this study confirmed and
quantified what has previously been proposed: that LADs [lamina-associated
domains] are genuine repressive, but heterogeneous, nuclear environments,
and that the interplay of promoter-intrinsic properties (e.g., bound
transcription factors) and local chromatin context determines final
transcriptional output ... Modeling generally correlates the most repressive
parts of LADs as those with the highest enrichment in lamin binding,
implying tightest attachment to the lamina. As may be expected, the most
transcriptionally permissive regions of LADs are generally enriched for
active histone modifications.” (Sexton 2019, doi:10.1016/j.tcb.2019.06.001).
“The nucleolus is the largest and most studied nuclear body, but its role in
nuclear function is far from being comprehensively understood. Much work on
the nucleolus has focused on its role in regulating RNA polymerase I (RNA
Pol I) transcription and ribosome biogenesis; however, emerging evidence
points to the nucleolus as an organizing hub for many nuclear functions,
accomplished via the shuttling of proteins and nucleic acids between the
nucleolus and nucleoplasm. Here, we discuss the cellular mechanisms affected
by shuttling of nucleolar components, including the 3D organization of the
genome, stress response, DNA repair and recombination, transcription
regulation, telomere maintenance, and other essential cellular functions”
(Iarovaia, Minina, Sheval 2019, 10.1016/j.tcb.2019.04.003).
“The nucleolus organizes the adjacent chromatin into a large-scale
repressive hub underlying the spatial segregation of active and repressive
chromatin compartments. Interphase chromosomes attach to the nucleolus via
nucleolus-associated domains (NADs). Sequestration of proteins within the
nucleolus allows their concentration to decrease sharply within other
cellular compartments. Perinucleolar regions contain a recombination
compartment; they also participate in allelic exclusion and X chromosome
silencing” (Iarovaia, Minina, Sheval 2019, 10.1016/j.tcb.2019.04.003).
“The genome of eukaryotic cells is organized into chromatin, a nuclear
complex comprising DNA, RNA, and associated proteins. Chromatin organization
displays hierarchical levels ranging from the basic repeated unit, the
nucleosome, to higher-level structures. The nucleosome is composed of a
core particle with ~147 base pairs of double-stranded DNA wrapped around
histone proteins with linker DNA joining core nucleosomal units. The
chromatin filament further coils and compacts DNA to reach higher-order
states with interacting chromatin loops and topologically associating
domains (TADs). Histones come as distinct variants that undergo
posttranslational modification (PTM) to provide modularity within core
particles. Histone chaperones, chromatin remodelers, and histone- and
DNA-modifying enzymes, along with PTM readers, transcription factors, and
RNA, generate specialized genomic domains for a versatile chromatin
landscape. Centromeres, telomeres, and regulatory elements display unique
nucleosome composition and structure. Modulation at each level enables
chromatin-based information to vary in order to respond to different signals
for numerous gene regulatory functions. This defines chromatin plasticity as
a means to generate a diversity of properties for each cell type during
development and also when cells face different environmental factors,
genotoxic insults, metabolic changes, senescence, disease, and even death”
(doi:10.1126/science.aat8950).
“It is reasonable to assume that chromatin in a typical human cell
consists of several thousands of different proteins”. Further, “in
chromatin, there may be several tens of thousands of distinct pairwise
protein-protein interactions, of which we currently know only a tiny
fraction. If we also consider the many non-coding RNA molecules that are
being discovered as part of chromatin, it becomes clear that chromatin is
an incredibly complex macromolecule” (Steensel 2011). This is a lot to
comprehend, when you consider that chromatin structure has a major and
intricate effect upon gene expression.
From an article detailing the remarkably complexity of factors affecting
chromatin structure:
“Different regulatory factors establish preferential contacts at different
scales. These range from close cis interactions such as promoter-gene body;
to long-range TAD- [topologically associated domain-] delimited contacts
such as those between enhancers and promoters and TF [transcription-factor]
binding sites; and finally, to very long-range contacts involving promoters,
Polycomb [proteins], heterochromatin regions, and a subset of TF binding
sites” (Bonev, Cohen, Szabo et al. 2017, doi:10.1016/j.cell.2017.09.043).
“Chromatin is a mighty consumer of cellular energy generated by metabolism.
Metabolic status is efficiently coordinated with transcription and
translation, which also feed back to regulate metabolism. Conversely,
suppression of energy utilization by chromatin processes may serve to
preserve energy resources for cell survival. Most of the reactions involved
in chromatin modification require metabolites as their cofactors or
coenzymes. Therefore, the metabolic status of the cell can influence the
spectra of posttranslational histone modifications and the structure,
density and location of nucleosomes, impacting epigenetic processes. Thus,
transcription, translation, and DNA/RNA biogenesis adapt to cellular
metabolism. In addition to dysfunctions of metabolic enzymes, imbalances
between metabolism and chromatin activities trigger metabolic disease and
life span alteration”
(Suganuma and Workman 2018, doi:10.1146/annurev-biochem-062917-012634).
“In higher eukaryotes, many genes are regulated by enhancers that are
104–106 base pairs (bp) away from the promoter. Enhancers contain
transcription-factor-binding sites (which are typically around 7–22 bp), and
physical contact between the promoters and enhancers is thought to be
required to modulate gene expression ... We find that highly punctate
contacts occur between enhancers, promoters and CCCTC-binding factor (CTCF)
sites and we show that transcription factors have an important role in the
maintenance of the contacts between enhancers and promoters. Our data show
that interactions between CTCF sites are increased when active promoters and
enhancers are located within the intervening chromatin. This supports a
model in which chromatin loop extrusion1 is dependent on cohesin loading at
active promoters and enhancers, which explains the formation of
tissue-specific chromatin domains without changes in CTCF binding”
(Hua, Badat, Hanssen et al. 2021, doi:10.1038/s41586-021-03639-4).
“Non-histone chromatin proteins such as the Heterochromatin Protein 1 (HP1)
proteins are critical regulators of transcription, contributing to gene
regulation through a variety of molecular mechanisms ... Given the presence
of multiple HP1 family members within a genome, HP1 proteins can have unique
as well as shared functions ... In Drosophila melanogaster, as in
other species, HP1 proteins can act as transcriptional repressors and
activators. The available data reveal that the precise impact of HP1
proteins on gene expression is highly context dependent, on the specific HP1
protein involved, on its protein partners present, and on the specific
chromatin context the interaction occurs in. As a group, HP1 proteins
utilize a variety of mechanisms to contribute to transcriptional regulation,
including both transcriptional (i.e. chromatin-based) and
post-transcriptional (i.e. RNA-based) processes. Despite extensive studies
of this important protein family, open questions regarding their functions
in gene regulation remain, specifically regarding the role of hetero-versus
homodimerization and post-translational modifications of HP1 proteins”
(Schoelz and Riddle 2022, doi:10.1186/s13072-022-00453-8).
-
Chromatin condensation and decondensation
- At a crude level: expression is largely, or at least relatively, repressed in highly condensed chromatin (heterochromatin) and much more freely allowed in decondensed chromatin (euchromatin). Many proteins (including the histones forming nucleosome core particles) and RNAs play a role in structuring chromatin. “It has been clear that the plasticity of and the dynamics of higher-order chromatin compaction are key regulators of transcription and other biological processes inherent to DNA” (Li and Reinberg 2011). “Chromatin regulates remarkably diverse processes in eukaryotic organisms, from development and disease progression to cognition and aging” (Zhang and Pugh 2011).
- More nuanced models are emerging where at least several broadly classified types of chromatin are being recognized (Steensel 2011).
- Various studies “have indicated that organisms alter how they package their DNA as they age. DNA doesn’t just float free. Our cells wrap their genetic material around proteins to form chromatin. Young, vigorous cells typically scrunch some of their chromatin into an orderly arrangement known as heterochromatin”. In a new study comparing older and younger people, “researchers found less heterochromatin in the older group, suggesting that their DNA had become disorganized with age”. “‘This study provides evidence that abnormal chromatin structure ... is likely a major contributing factor to premature aging characteristic of the genetic disorder Werner syndrome,’ says molecular biologist Robert Brosh of the National Institute on Aging in Bethesda, Maryland, who wasn’t connected to the research. In addition, he says, the work suggests that ‘defective chromatin organization may underlie normal aging as well’” (Leslie 2015, doi:10.1126/science.aab2575).
- “Constitutive heterochromatin has traditionally been viewed as a highly-stable structure that represses the transcription and recombination of repetitive DNA elements. However, recent studies have demonstrated that constitutive heterochromatin domains are also highly dynamic. The function of such dynamics is only beginning to be appreciated ... and it might be part of the cellular response to outside stimuli by modifying chromatin structure to cushion against adverse effects. The silencing of gene expression by heterochromatin in a sequence-independent manner makes heterochromatin formation one of the most versatile forms of epigenetic changes. Adaptive changes of heterochromatin in response to numerous stresses take place in diverse species from yeasts to humans. Because a crucial step in tumor development is the inactivation of tumor-suppressor genes, the discoveries of epigenetic inactivation phenomena in different systems provide invaluable clues for studying the adaptation of tumor cells and designing new strategies to counteract such effects” (Wang, Jia and Jia 2016, doi:10.1016/j.tig.2016.02.005).
- “In mammals, chromatin organization undergoes drastic reprogramming after fertilization ... We found that oocytes in metaphase II show homogeneous chromatin folding that lacks detectable topologically associating domains (TADs) and chromatin compartments. Strikingly, chromatin shows greatly diminished higher-order structure after fertilization. Unexpectedly, the subsequent establishment of chromatin organization is a prolonged process that extends through preimplantation development, as characterized by slow consolidation of TADs and segregation of chromatin compartments. The two sets of parental chromosomes are spatially separated from each other and display distinct compartmentalization in zygotes. Such allele separation and allelic compartmentalization can be found as late as the 8-cell stage. Finally, we show that chromatin compaction in preimplantation embryos can partially proceed in the absence of zygotic transcription and is a multi-level hierarchical process. Taken together, our data suggest that chromatin may exist in a markedly relaxed state after fertilization, followed by progressive maturation of higher-order chromatin architecture during early development” (Du, Zheng, Huang et al. 2017, doi:10.1038/nature23263).
-
Nucleosomes play a central role in the packaging of chromatin and
in the accessibility of DNA by protein binding factors.
- DNA wraps around histone core particles, and the resulting nucleosomes can form the tightly packed arrays characteristic of condensed chromatin.
- Where DNA enters and exits a nucleosome spool, a linker histone — distinct from the histones constituting the spools — can bind the entering and exiting DNA together, thereby “sealing” the DNA to the spool, rendering the DNA less accessible, stabilizing the spool, and (when linker histones are present along a considerable length of the chromosome) conducing to the formation of compact nucleosome arrays and chromosome condensation.
- “It is being increasingly realized that nucleosome organization on DNA crucially regulates DNA-protein interactions and the resulting gene expression. While the spatial character of the nucleosome positioning on DNA has been experimentally and theoretically studied extensively, the temporal character is poorly understood. Accounting for ATPase activity and DNA-sequence effects on nucleosome kinetics, we develop a theoretical method to estimate the time of continuous exposure of binding sites of non-histone proteins (e.g. transcription factors and TATA binding proteins) along any genome. Applying the method to Saccharomyces cerevisiae, we show that the exposure timescales are determined by cooperative dynamics of multiple nucleosomes, and their behavior is often different from expectations based on static nucleosome occupancy. Examining exposure times in the promoters of GAL1 and PHO5, we show that our theoretical predictions are consistent with known experiments” (Parmar, Das and Padinhateeri 2016, doi:10.1093/nar/gkv1153).
-
Histone chaperones. “Histone chaperones, which are proteins
that escort histones throughout their cellular life, are key actors in
all facets of histone metabolism; they regulate the supply and dynamics
of histones at chromatin for its assembly and disassembly. Histone
chaperones can also participate in the distribution of histone
variants, thereby defining distinct chromatin landscapes of importance
for genome function, stability, and cell identity”. “Histone
chaperones provide interfaces that allow their recruitment to
particular genomic loci or that link to specific biological processes”.
(Gurard-Levin, Quivy and Almouzni 2014,
doi:10.1146/annurev-biochem-060713-035536). You will find histone
chaperones mentioned under other various other headings of this
document.
- “Histone chaperones can handle and buffer histones displaced ahead of the polymerase, thereby functioning as a so-called histone sink. Indeed, several histone chaperones have been implicated in accepting H2A–H2B dimers to facilitate transcription factor binding ... Following the passage of RNA Pol II, the reassembly of nucleosomes restores the chromatin structure, preventing cryptic transcription ... Thus, similarly to replication and repair, transcription represents another transient disruption to the chromatin organization and another window of opportunity to either maintain or alter the chromatin landscape” (Gurard-Levin, Quivy and Almouzni 2014, doi:10.1146/annurev-biochem-060713-035536).
-
Chromatin remodeling proteins — many families and subfamilies of
them — also play a decisive role in structuring chromatin.
“The TGF-β/BMP signaling superfamily plays critical roles in regulating a
wide range of physiological and pathological processes. A recent study on
Tgfbr1 function during mouse embryonic development found that, in the
absence of this gene, the primordium of the external genitalia changes
its fate, forming an additional set of hindlimbs and resulting in the
so-called “six-legged” mice. Analysis of these embryos demonstrated that
Tgfbr1 induces global remodeling of chromatin accessibility, establishing
distinct regulatory elements that other morphogenetic factors use to
drive either hindlimb or genital development. While global changes in
chromatin structure have been previously associated with TGF-β signaling,
the alterations seen in six-legged embryos differ significantly from
previously described mechanisms. In this review, I discuss these
distinctions and underscore the importance of studying biological
processes within their natural tissue context, where essential tissue
interactions are preserved”
(Mallo 2025, doi:10.1002/bies.70013).
- In addition to histones, numerous proteins can bind to chromatin and shape its architecture, bending its DNA (as is required for the start of transcription), or joining more or less distantly separated sites together so as to form loops, or bringing extended lengths of DNA side by side.
-
It is thought that a particular “high mobility group” protein
(HMGB1) binds to DNA at its core histone entry and exit points and
introduces a bend in the DNA. This has the effect of loosening the
DNA from the nucleosome core, making the DNA more accessible to
transcription factors and other regulatory molecules.
- On the other hand, the bending of DNA by HMGB1 can promote chromatin compaction (Luijsterburg, White, Driel and Dame 2008).
- The bending and loosening of DNA from the nucleosome core might also promote nucleosome remodeling (Luijsterburg, White, Driel and Dame 2008).
- There is no hard-and-fast distinction between remodeling proteins that more or less directly affect chromatin structure (and therefore also gene expression), on the one hand, and proteins that apply histone modifications, which can also have major effects on chromatin structure — for example, by altering the relationship between neighboring nucleosomes or by lowering or increasing a remodeling protein’s affinity for a particular site.
- Here, as elsewhere, context matters. By facilitating the formation of heterochromatin, HP1 (heterochromatin protein 1) has been primarily associated with gene repression. However, in the context of euchromatin it is being found to play positive roles in gene expression. For example, by recruiting a histone chaperone complex to active genes and linking it to RNA polymerase, it can play an important part in transcription elongation (Kwon, Florens, Swanson et al. 2010).
- Remodeling proteins are themselves subject to modification by the addition of various chemical groups. The addition of a phosphoryl group is a common means by which the activity of a regulatory protein is modified. For example, HP1 helps to compact chromatin into heterochromatin by binding to certain histone modifications. However, the phosphorylation of a particular part of HP1 leads it to dissociate from the histone. As another example of such modification — in this case, connected with spatial organization of the nucleus and long noncoding RNAs — see the item about PC2 under “Long noncoding RNAs” below.
- “We identify a new property of the human HP1α protein: the ability to form phase-separated droplets. While unmodified HP1α is soluble, either phosphorylation of its N-terminal extension or DNA binding promotes the formation of phase-separated droplets. Phosphorylation-driven phase separation can be promoted or reversed by specific HP1α ligands. Known components of heterochromatin such as nucleosomes and DNA preferentially partition into the HP1α droplets, but molecules such as the transcription factor TFIIB show no preference ... Both unmodified and phosphorylated HP1α induce rapid compaction of DNA strands into puncta, although with different characteristics ... an HP1α mutant incapable of phase separation in vitro forms smaller and fewer nuclear puncta than phosphorylated HP1α. These findings suggest that heterochromatin-mediated gene silencing may occur in part through sequestration of compacted chromatin in phase-separated HP1 droplets, which are dissolved or formed by specific ligands on the basis of nuclear context” (Larson, Elnatan, Keenen et al. 2017, doi:10.1038/nature22822). See also Ribonucleoprotein phase transitions below.
- Remodeling proteins can have a direct effect on gene expression simply by outcompeting transcription factors for DNA binding sites (Luijsterburg, White, Driel and Dame 2008).
- Another form of competition: An HMG (high mobility group) protein competes with linker histones for binding to linker DNA, thereby conducing to destabilization of higher-order chromatin structure and transcriptional activation. But compare the ability of HMG proteins to facilitate chromatin compaction.
- One of countless examples of particular molecular roles (from yeast): “We find a substantial influence of [chromatin remodeling complex] INO80 on nucleosome dynamics and gene expression during stress induced transcription. Transcription induced by osmotic stress leads to genome-wide remodeling of promoter proximal nucleosomes. INO80 function is required for timely return of evicted nucleosomes to the 5' end of induced genes. Reduced INO80 function in Arp8-deficient cells leads to correlated prolonged transcription and nucleosome eviction. INO80 and the related complex SWR1 regulate incorporation of the H2A.Z isoform at promoter proximal nucleosomes. However, H2A.Z seems not to influence osmotic stress induced gene regulation. Furthermore, we show that high rates of transcription promote INO80 recruitment to promoter regions, suggesting a connection between active transcription and promoter proximal nucleosome remodeling. In addition, we find that absence of INO80 enhances bidirectional promoter activity at highly induced genes and expression of a number of stress induced transcripts” (Klopf, Schmidt, Clauder-Münster et al. 2017, doi:10.1093/nar/gkw1292)
-
Polycomb repressor complexes (PRC1 and PRC2)
These complexes constitute just one of many types of chromatin
remodeling proteins. They are referred to throughout this
document; no attempt is made to focus their treatment here. As
their name implies, they have long been associated with repression
of gene expression by facilitating the formation of heterochromatin
in cooperation with other factors. However, as with nearly all
elements of gene regulation, the more we learn about these
complexes, the more contextually dependent and varied their
activity becomes.
“Polycomb repressive complex 2 (PRC2) is a multisubunit protein
complex essential for the development of multicellular organisms.
Recruitment of PRC2 to target genes, followed by deposition and
propagation of its catalytic product histone H3 lysine 27
trimethylation (H3K27me3), are key to the spatiotemporal control of
developmental gene expression. Recent breakthrough studies have
uncovered unexpected roles for substoichiometric PRC2 subunits in
these processes. Here, we elaborate on how the facultative PRC2
subunits regulate catalytic activity, locus-specific PRC2 binding, and
propagation of H3K27me3, and how this affects chromatin structure,
gene expression, and cell fate”
(van Mierlo, Veenstra Vermeulen et al., 2019,
doi:10.1016/j.tcb.2019.05.004).
“PRC2 can associate with a multitude of facultative subunits resulting
in at least two different multimeric configurations called PRC2.1 and
PRC2.2. However, the exact interplay between the two subcomplexes
requires further study. PRC2.1 and PRC2.2 are recruited to target
genes via distinct mechanisms and have divergent roles in gene
silencing, but their combined action together with PRC1 is required
for Polycomb function. Changes of cellular state, such as during the
differentiation of embryonic stem cells, can evoke architectural and
functional changes in the PRC2 subcomplexes”
(van Mierlo, Veenstra Vermeulen et al., 2019,
doi:10.1016/j.tcb.2019.05.004).
“In developing embryos, dynamic PRC binding contributes to Hox
regulation first by directly binding genes to keep them inactive and
prevent them from contacting active enhancers. Second, PRC‐dependent
chromatin contacts establish a 3D chromatin architecture that preset
enhancer‐promoter physical proximity, favoring gene activation upon
loss of PRC binding” (Table of Contents blurb for
Gentile and Kmita 2020, doi:10.1002/bies.201900249).
“Although polycomb repressive complex 2 (PRC2) is now recognized as an
RNA-binding complex, the full range of binding motifs and why PRC2–RNA
complexes often associate with active genes have not been elucidated.
Here, we identify high-affinity RNA motifs whose mutations weaken PRC2
binding and attenuate its repressive function in mouse embryonic stem
cells. Interactions occur at promoter-proximal regions and frequently
coincide with pausing of RNA polymerase II (POL-II). Surprisingly,
while PRC2-associated nascent transcripts are highly expressed,
ablating PRC2 further upregulates expression via loss of pausing and
enhanced transcription elongation. Thus, PRC2-nascent RNA complexes
operate as rheostats to fine-tune transcription by regulating
transitions between pausing and elongation, explaining why PRC2–RNA
complexes frequently occur within active genes. Nascent RNA also
targets PRC2 in cis and downregulates neighboring genes. We propose a
unifying model in which RNA specifically recruits PRC2 to repress
genes through POL-II pausing and, more classically, trimethylation of
histone H3 at Lys27”
(Rosenberg, Blum, Kesner et al. 2021, doi:10.1038/s41594-020-00535-9).
[Interweaving roles of various epigenetic factors:]
“Distinct classes of cofactor proteins are known to regulate the
functional activity and interplay of [PRC1 and PRC2] ... Furthermore,
PRC2 cofactors like AEBP2 and JARID2 also play a role in mediating the
cross-talk between different histone posttranslational modifications
and PRC2 recruitment and activity — a function that is important for
the regulated control of gene expression. PRC1 is an E3 ubiquitin
ligase responsible for the monoubiquitination of histone H2A
(H2AK119ub1), a histone mark recognized by PRC2 and linked to its
genomic recruitment ... We find that JARID2 recognizes both the
ubiquitin moiety in H2AK119ub1 and the conserved histone H2A-H2B
acidic patch. We also observe that the tandem zinc fingers of AEBP2
interact with ubiquitin and the histone H2A-H2B surface on the other
side of the nucleosome. Biochemical assays show a secondary activation
of PRC2 by JARID2 and AEBP2 on H2AK119ub1-containing nucleosomes
besides the primary [polycomb protein] EED-mediated allosteric
activation of PRC2 by methylated JARID2. Furthermore, we also find
that the joint presence of JARID2 and AEBP2 partially reduces the
inhibition of PRC2 methyltransferase activity by the transcriptionally
active histone posttranslational modifications H3K4me3 and H3K36me3.
Cryo-EM visualization of PRC2 that contains JARID2 and AEBP2
interacting with a H3K4me3-containing nucleosome shows the coexistence
of states in which the histone H3 tail is either absent or engaged and
reaching the catalytic site in PRC2, which provides a physical basis
for the partial activity of the complex on H3K4me3-containing
nucleosomes. [In sum:] Our studies indicate that cofactors JARID2 and
AEBP2 play a crucial role in both the recruitment and activation of
PRC2 through their recognition of H2AK119ub1, which is generated by
PRC1. Additionally, our work suggests that JARID2 and AEBP2 are likely
to play a key role in regulating PRC2 activity on genomic regions with
active transcription marks”
(Kasinath, Beck, Sauer et al. 2021, doi:10.1126/science.abc3393).
“Modulation of chromatin structure and/or modification by Polycomb
repressive complexes (PRCs) provides an important means to partition
the genome into functionally distinct subdomains and to regulate the
activity of the underlying genes. Both the enzymatic activity of PRC2
and its chromatin recruitment, spreading, and eviction are exquisitely
regulated via interactions with cofactors and DNA elements (such as
unmethylated CpG islands), histones, RNA (nascent mRNA and long
noncoding RNA), and R-loops. PRC2-catalyzed histone H3 lysine 27
trimethylation (H3K27me3) is recognized by distinct classes of
effectors such as canonical PRC1 and BAH module-containing proteins
(notably BAHCC1 in human). These effectors mediate gene silencing by
different mechanisms including phase separation-related chromatin
compaction and histone deacetylation”
(Guo, Zhao and Wang 2021, doi:10.1016/j.tig.2020.12.006).
- Polycomb repressor complexes (PRCs) play a particularly strong role in pluripotent and stem cells, as well as cancer cells. In an apparent paradox, their silencing activity in embryonic stem cells “can be accompanied by active chromatin and primed RNA polymerase II”. PRCs target different variants of RNA polymerase II, with the variants distinguished by which serine residues are phosphorylated on the C-terminal tail of the polymerase. Gene silencing occurs in some cases, but activation can occur in others. In particular, the active state alternates with a repressive state as the phosphorylation of RNA polymerase II changes. This fluctuation is thought to vary across different gene targets of PRC, leading to different gene expression levels (Brookes, de Santiago, Hebenstreit et al. 2012).
- Polycomb complexes have been primarily studied in relation to early development. But it has now been shown that PRC2 “is required for the proper control of cell fate decisions in the adult intestinal epithelium”: “Epigenetic control of gene expression in adult tissues is crucial to maintain organ function and homeostasis ... chromatin repressive complex PRC2 controls the equilibrium between secretory and absorptive fates in the intestine. PRC2 controls proliferation of cells within the crypt and at the same time represses the transcription factor Atoh1, thus favoring the generation of enterocytes versus secretory cell types in the adult intestine” (Vizán, Beringer, and Croce 2016, doi:10.15252/embj.201695694).
- “In addition to four core subunits, PRC2 comprises multiple accessory subunits that vary in their composition during cellular differentiation and define two major holo-PRC2 complexes: PRC2.1 and PRC2.2. PRC2 binds to RNA, which inhibits its enzymatic activity, but the mechanism of RNA-mediated inhibition of holo-PRC2 is poorly understood. Here we present in vivo and in vitro protein-RNA interaction maps and identify an RNA-binding patch within the allosteric regulatory site of human and mouse PRC2, adjacent to the methyltransferase center. RNA-mediated inhibition of holo-PRC2 is relieved by allosteric activation of PRC2 by H3K27me3 and JARID2-K116me3 peptides. Both holo-PRC2.1 and holo-PRC2.2 bind RNA, providing a unified model to explain how RNA and allosteric stimuli antagonistically regulate the enzymatic activity of PRC2” (Zhang, McKenzie, Warneford-Thomson et al. 2019, doi:10.1038/s41594-019-0197-y).
- PRC2 and miRNA interdependence. “We found that Polycomb repressive complex 2 (PRC2) and its associated histone mark, H3K27me3, is enriched at hundreds of miRNA-repressed genes. We show that these genes are directly repressed by PRC2 and constitute a significant proportion of direct PRC2 targets. For just over half of the genes corepressed by PRC2 and miRNAs, PRC2 promotes their miRNA-mediated repression by increasing expression of the miRNAs that are likely to target them. miRNAs also repress the remainder of the PRC2 target genes, but independently of PRC2. Thus, miRNAs post-transcriptionally reinforce silencing of PRC2-repressed genes that are inefficiently repressed at the level of chromatin, by either forming a feed-forward regulatory network with PRC2 or repressing them independently of PRC2.” (Shivram, Le and R. Iyer 2019, doi:10.1101/gr.238311.118)
-
ATP-dependent chromatin remodeling enzymes
These enzymes use energy from ATP to remodel the chromosome-histone
complexes that constitute nucleosomes. The remodeling can have
dramatic effects upon gene expression, in part (and only in part)
because DNA tightly bound to a nucleosome is less accessible for
transcription than more loosely bound or nucleosome-free DNA. (For
some of the gene regulation implications, see all the preceding
topics relating to nucleosomes and histones.) The SWI/SNF and RSC
families of related proteins make up two groups of ATP-dependent
remodeling enzymes.
“Together, the different subfamilies of chromatin-remodeling
enzymes catalyze a broad range of chromatin transformations that
includes sliding the histone octamer across the DNA, changing the
conformation of nucleosomal DNA, and changing the composition of
the histone octamer. These biochemical activities are remarkable
given the underlying mechanistic challenges. The substrate, a
nucleosome, is structurally complex and contains DNA tightly bound
to the histone octamer. Somehow, chromatin-remodeling enzymes have
to disrupt DNA-histone interactions while contending with and
leveraging the structural constraints placed by the histone
octamer” (Narlikar, Sundaramoorthy and Owen-Hughes 2013).
- Researchers mapped the genome-wide binding of mammalian Brg1, Snf2h, and Chd4 ATPases, which are at the core of multiple remodeling complexes. They found about “40,000 sites occupied by each remodeler, with the majority of the binding sites in the promoter regions and gene bodies. ... Most remarkably, they discovered that binding sites between remodelers showed a high degree of overlap, with more than 50% of sites being shared by all three remodelers and an even greater proportion shared by at least two” (Varga-Weisz 2014).
- According to the same study, “each remodeler renders some sites accessible while closing others ... with evidence for synergistic and opposing actions by distinct remodelers. This study illustrates that multiple remodelers act over the same sites to shape chromatin and emphasizes the need to view chromatin dynamics as the action of multiple factors, possibly successively, over the same site (Varga-Weisz 2014).
- The collection of subunits in human ATP-dependent remodeling complexes varies according to tissue type. Also, “mutations in components of human remodeling complexes have now been identified at high frequencies in human cancers” This may involve a role of the complexes in genome instability. “However, the specificity with which inactivation of different subunits [of the remodeling complexes] affects different types of cancer suggests more complex tissue specific modes of action (Narlikar, Sundaramoorthy and Owen-Hughes 2013).
- Some ATP-dependent chromatin remodelers such as the SWI/SNF family act to randomize nucleosome positions by sliding nucleosomes along DNA. Other remodelers slide nucleosomes in order to achieve equal spacing between them, thereby facilitating higher-order chromatin compaction (Luijsterburg et al. 2008).
- “The nature of the alteration to chromatin occurring at sites of SWI/SNF recruitment has not been characterized in all cases. However, examples exist to support nucleosome repositioning, disruption, and histone removal in different contexts.” (Narlikar, Sundaramoorthy and Owen-Hughes 2013).
- RSC enzymes appears to play a role in nucleosome-removal, helping to maintain nucleosome-depletion in the region upstream from gene promoters, and perhaps in other regulatory regions as well. This may occur in conjunction with bound transcription factors (Narlikar, Sundaramoorthy and Owen-Hughes 2013).
- “RSC and SWI/SNF can move two nucleosomes into such close proximity that DNA is unwound from the histone octamer at the interface of the two nucleosomes. ... The [enzyme-]bound nucleosome appears to be used as a ram, destabilizing nucleosomes that it collides with. As a result, it would be expected that a single nucleosome would not be removed from DNA as effectively as one surrounded by neighbors. Consistent with this expectation, RSC removes nucleosomes more effectively from multinucleosome templates” (Narlikar, Sundaramoorthy and Owen-Hughes 2013).
- RSC and some subunits of SWI/SNF have been shown to bind to histone tails, and this is affected by histone tail modifications. Apparently the binding can change the conformation of at least some of the enzymes, suggesting that the latter do not have a fixed effect, but rather “a change in the type of interaction between a remodeling enzyme and nucleosomes can alter the outcome of remodeling” (Narlikar, Sundaramoorthy and Owen-Hughes 2013).
- Some ATP-dependent chromatin remodeling enzymes have special capabilities for facilitating histone exchange. For example, the Swr1 complex replaces histone H2A/H2B dimers with H2AZ/H2B dimers. (See Histone variants under Nucleosome remodeling above.) “There are at least three ways to influence the presence of a histone variant: targeted incorporation illustrated by Swr1, targeted removal as illustrated by Ino80, and increased exchange as illustrated by Fun30”. In the case of histone variant H2A.Z, its post-translational modifications may help to regulate its distribution (Narlikar, Sundaramoorthy and Owen-Hughes 2013).
- ATP-dependent chromatin-remodeling enzymes may achieve their tasks in conjunction with protein chaperone molecules. For example, in humans the ATRX enzyme associates with a chaperone specific to the H3.3 histone variant. ATRX then apparently couples dissociation of nucleosomes with H3.3-enriched reassembly (Narlikar, Sundaramoorthy and Owen-Hughes 2013).
- Unsurprisingly, given all the above, RSC and SWI/SNF have been shown to play roles in the regulation of transcriptional elongation. “It is tempting to speculate that this role involves assisting the removal of histones from DNA during transcription by RNA polymerase” (Narlikar, Sundaramoorthy and Owen-Huges 2013).
-
Chromatin breathing
- Pluripotent stem cells exhibit what has been called “chromatin breathing”, which is marked by the rapid exchange of certain histones and other proteins. That is, the cycling (binding and release) of these proteins on chromatin is very rapid — for example, a few seconds per cycle for the linker histone H1, and slightly more than a minute for the H2B and H3 core histones. This “hyperdynamic" chromatin, with certain structuring proteins only loosely bound, appears not only to be characteristic of pluripotent cells, but also a prerequisite for their subsequent differentiation. After differentiation, the rate of molecular exchange slows down, coincident with the relative inactivation of a substantial portion of the genome in a condensed, or heterochromatic, form (Meshorer, Yellajoshula, George et al. 2006; Zwaka 2006).
- This hyperdynamic binding of structural proteins is correlated with vibrational, or rhythmic, movements of chromatin. “We show that pluripotency is associated with a highly discrete, energy-dependent frequency of chromatin movement that we refer to as a ‘breathing’ state. We find that this ‘breathing’ state is strictly dependent on the metabolic state of the cell and is progressively silenced during differentiation, thus presumably representing a hallmark of pluripotency maintenance”. The vibration frequency is 10 – 100 Hz. It is thought that such movement helps to maintain the chromatin of pluripotent cells in an open or uncondensed state (Hind, Cardarelli, Chen et al. 2012).
-
Chromatin-associated RNAs
“Chromatin-associated RNAs regulate facultative and constitutive
heterochromatin. RNA can recruit, stabilize, inhibit activity, or
prevent spread of heterochromatin proteins. Chromatin-associated RNAs
regulate heterochromatin by both cis and trans mechanisms.
Small RNAs or long non-coding RNAs recruit heterochromatin factors”
(Johnson and Straight 2017, doi:10.1016/j.ceb.2017.05.004).
- DNA methylation helps determine protein affinities for potential binding sites. Methylation is often associated with chromatin compaction, and demethylation with open chromatin. On the general topic of DNA methylation, see DNA methylation under PRE-TRANSCRIPTIONAL DECISION-MAKING above.
-
Topoisomerases
Topoisomerases are enzymes that cut one or both strands of the DNA
double helix and then — after topological changes are made in the DNA —
reconnects the strands. By this means the double helix can be wound
more or less tightly, and knots in the DNA can be managed.
- “DNA topoisomerases are thought to facilitate transcription by removing excess topological strain induced by the tracking of the polymerase. A study in Saccharomyces cerevisiae deficient for topoisomerases I and II has now suggested that in vivo [topoisomerases] are also involved in gene activation. Genes particularly affected in topoisomerase mutants have features associated with highly regulated transcription, such as a TATA box, which is indicative of a repressible and/or inducible mode of transcription. For the gene PHO5 ... topoisomerases are required for transcription factor binding” (Stower 2013).
- “We show that topoisomerase I activity is directly required for efficient nucleosome disassembly at gene promoter regions. Lack of topoisomerase activity results in increased nucleosome occupancy, perturbed histone modifications and reduced transcription from these promoters. Strong correlative evidence suggests that topoisomerase I cooperates...in nucleosome disassembly. Our study links topoisomerase activity to the maintenance of open chromatin and regulating transcription in vivo” (Durand-Dubief, Persson, Norman et al. 2010).
- Some gene expression in neurons needs to occur rapidly in response to sensory stimuli. Many factors required for this high-level, “activity-dependent” gene expression are already in place (“poised”) before stimulation. The question is what holds back expression before a stimulus, and what releases the transcriptional process after the stimulus. Now evidence has been produced that “activity-regulated genes are maintained in a state of high torsional stress prior to stimulation such that supercoiling of the DNA keeps RNAPII [RNA polymerase II] from extending into gene bodies ... upon neuronal depolarization, activation of Topoisomerase IIB leads to DNA double-stranded breaks (DSBs) within the promoters, thus allowing the DNA to unwind and RNAPII to productively elongate through gene bodies”. It is suggested that “topoisomerase pathways may play a particularly important role in transcriptional regulation in the brain” (Sharma, Gabel and Greenberg 2015, doi:10.1016/j.cell.2015.06.009).
This topic was only recently added, and should cover all the different
“nuclear bodies” — membraneless structures found in the cell nucleus. While
the functions of these bodies are in many cases unclear, they are deeply
connected to gene expression. The following is merely a placeholder for a
wide-ranging set of regulatory functions related to nuclear bodies.
- “Recent studies showed that active chromatin regions are associated with nuclear speckles (NSs), a type of NBs [nuclear bodies] involved in RNA processing ... Using mouse hepatocytes as the model, we knocked down SRRM2, a core protein component scaffolding NSs ... We found that Srrm2 depletion disrupted the NSs and changed the expression of 1282 genes. The intra-chromosomal interactions were decreased in type A (active) compartments and increased in type B (repressive) compartments. Furthermore, upon Srrm2 knockdown, the insulation of TADs [topologically associated domains] was decreased specifically in active compartments, and the most significant reduction occurred in A1 sub-compartments ... We show that disruption of NSs by Srrm2 knockdown causes a global decrease in chromatin interactions in active compartments, indicating critical functions of NSs in the organization of the 3D genome” (Hu, Lv, Yan and Wen 2019, doi:10.1186/s13072-019-0289-2).
“Every one of the better-understood epigenetic information carriers
exhibits crosstalk with every one of the other carriers. Cytosine
modifications directly affect nucleosome positioning and recruit
chromatin-modifying complexes, and conversely histone modifications can
affect recruitment of cytosine methylases and demethylases. Small RNAs,
including short interfering RNAs (siRNAs) and piRNAs, and long RNAs, such
as long intergenic noncoding RNAs (lincRNAs), can direct histone
modifications and cytosine methylation. Finally, chromatin structure and
DNA modifications affect transcription of small RNA and lincRNA-containing
loci” (Rando 2012).
This is a vast topic touching upon just about all aspects of biological
functioning. Here we barely allude to a few generalities.
Signaling processes play a central role in regulating gene expression.
They are a primary means by which gene expression can respond to, and be
properly calibrated to, external conditions — whether those conditions
occur within the larger cell or outside the cell. For example, a hormone
distributed via the blood stream may interact with a receptor at the cell
surface, which in turn may trigger a cascade of interactions within the
cell, culminating in transcription factors or other regulatory factors
coming to bear on DNA
- Signaling pathways were formerly thought of rather straightforwardly as consisting of a single, well-defined input, a linear series of interactions, and a well-defined output such as the production of a transcription factor. Now, however, individual proteins in signaling pathways are known to be capable of up to billions of distinct, functionally relevant states (Mayer, Blinov and Loew 2009) and to be involved in crosstalk with other pathways, so that attempts to tabulate the cross-signaling between just a few pathways yields a “horror graph” and it begins to look as though “everything does everything to everything” (Dumont, Pécasse and Maenhaut 2001).
- Regarding the temporal aspect of cell signaling: “Activation of a signalling network is dynamic, subject to receptor down-regulation and other forms of negative feedback adaptation. Thus, the magnitude of pathway activation typically peaks early before reaching a quasi-steady plateau...is it the steady state that matters most, or the peak? If the entire time course is important, how should [the mathematical modeler] weight the signalling magnitudes at different times? [A particular current conjecture] only highlights the fundamental difficulties we face when trying to simplify complex biology using mathematics” (Haugh 2012).
- “The bone morphogenetic protein (BMP) signaling pathway comprises multiple ligands and receptors that interact promiscuously with one another and typically appear in combinations ... Here, we show that the BMP pathway processes multi-ligand inputs using a specific repertoire of computations, including ratiometric sensing, balance detection, and imbalance detection. These computations operate on the relative levels of different ligands and can arise directly from competitive receptor-ligand interactions. Furthermore, cells can select different computations to perform on the same ligand combination through expression of alternative sets of receptor variants. These results provide a direct signal-processing role for promiscuous receptor-ligand interactions and establish operational principles for quantitatively controlling cells with BMP ligands. Similar principles could apply to other promiscuous signaling pathways”. Of course, all this is directly related to the regulation of gene expression: “Ligand combinations represent inputs to the pathway, which processes them through receptor-ligand interactions to control the expression level of down-stream target genes” (Antebi, Linton, Klumpe et al. 2017, doi:10.1016/j.cell.2017.08.015).
-
Protein modifications in general. It is not only the
chromatin-associated proteins whose modifications influence gene
expression. This is true of proteins in general, indicating how the
factors coming to bear on genes radiate in without any boundary.
- “Methylation of Lys and Arg residues on non-histone proteins has emerged as a prevalent post-translational modification and as an important regulator of cellular signal transduction mediated by the MAPK, WNT, BMP, Hippo and JAK–STAT signalling pathways. Crosstalk between methylation and other types of post-translational modifications, and between histone and non-histone protein methylation frequently occurs and affects cellular functions such as chromatin remodelling, gene transcription, protein synthesis, signal transduction and DNA repair. With recent advances in proteomic techniques, in particular mass spectrometry, the stage is now set to decode the methylproteome and define its functions in health and disease” (Biggar and Li 2015, doi:10.1038/nrm3915).
“Post-zygotic variation refers to genetic changes that arise in the soma of
an individual and that are not usually inherited by the next generation.
Although there is a paucity of research on such variation, emerging studies
show that it is common: individuals are complex mosaics of genetically
distinct cells, to such an extent that no two somatic cells are likely to
have the exact same genome. Although most types of mutation can be involved
in post-zygotic variation, structural genetic variants are likely to leave
the largest genomic footprint. Somatic variation has diverse physiological
roles and pathological consequences, particularly when acquired variants
influence the clonal trajectories of the affected cells”. “The concept that
the genome of the soma is not only variable but also changing over time is
not yet sufficiently recognized. Multiple lines of evidence reviewed here
suggest that the genetic composition of somatic cells making up a single
human soma is dynamic, evolving through time from conception to death”
(Forsberg, Gisselsson and Dumanski 2017, doi:10.1038/nrg.2016.145).
“Somatic mosaicism, the presence of more than one genotype in the somatic
cells of an individual, is a prominent phenomenon in the human central
nervous system. Forms of mosaicism include aneuploidies and smaller copy
number variants (CNVs), structural variants (SVs), mobile element
insertions, indels, and single nucleotide variants (SNVs). The developing
human brain exhibits high levels of aneuploidy compared to other tissues,
generating genetic diversity in neurons. Such aneuploidy was suggested to be
a natural feature of neurons, rather than a distinctive feature of
neurodegeneration. However, the frequency of aneuploidy in neurons has been
debated, with a separate study suggesting that aneuploidies occur in only
about 2.2% of mature adult neurons. They hence infer that such aneuploidy
could have adverse effects at the cellular and organismal levels.
Additionally, analysis of single cells from normal and pathological human
brains identified large, private, and likely clonal somatic CNVs in both
normal and diseased brains, with 3%–25% of human cerebral cortical nuclei
carrying megabase-scale CNVs and deletions being twice as common as
duplications. Given that CNVs often arise from nonhomologous recombination
and replication errors, their likely time of origin is during brain
development. However, when CNVs first arise in human brain development has
not yet been investigated”
(Sekar, Tomasini, Proukakis et al. 2020, doi:10.1101/gr.262667.120).
“We previously detected 200–400 mosaic SNVs [single nucleotide variants] per
cell in three human fetal brains (15–21 wk postconception). However,
structural variation in the human fetal brain has not yet been investigated.
Here, we discover and validate four mosaic structural variants (SVs) in the
same brains and resolve their precise breakpoints. The SVs were of kilobase
scale and complex, consisting of deletion(s) and rearranged genomic
fragments, which sometimes originated from different chromosomes. Sequences
at the breakpoints of these rearrangements had microhomologies, suggesting
their origin from replication errors. One SV was found in two clones, and we
timed its origin to ∼14 wk postconception. No large scale mosaic copy number
variants (CNVs) were detectable in normal fetal human brains, suggesting
that previously reported megabase-scale CNVs in neurons arise at later
stages of development. By reanalysis of public single nuclei data from adult
brain neurons, we detected an extrachromosomal circular DNA event. Our study
reveals the existence of mosaic SVs in the developing human brain, likely
arising from cell proliferation during mid-neurogenesis. Although relatively
rare compared to SNVs and present in ∼10% of neurons, SVs in developing
human brain affect a comparable number of bases in the genome (∼6200 vs.
∼4000 bp), implying that they may have similar functional consequences”
(Sekar, Tomasini, Proukakis et al. 2020, doi:10.1101/gr.262667.120).
(This could fall under a number of different headings.)
There are various ways in which one of the two alleles of a gene can
be expressed more than, or to the exclusion of, the other, with either
major or subtle consequences for the organism.
- X chromosome inactivation. (See X chromosome inactivation under “Negotiations Among Parents and Offspring” above.)
- Imprinting. (See “Imprinting” under “Negotiations Among Parents and Offspring” above.)
-
Autosomal monoallelic expression (MAE)
“MAE can be defined as a mosaic epigenetic inactivation of one allele
of an autosomal gene. Similarly to X-inactivation, some cells express
the paternal allele, while others cells of the same time in the same
individual express the maternal allele. The choice of active allele,
once made, appears to be maintained indefinitely. ... the epigenetic
allele choices are maintained genome-wide through dozens of cell
divisions”. It is estimated that between 10 and 20% of all mammalian
genes — a likely underestimate — are subject to monoallelic expression
(Savova, Vigneau and Gimelbrant 2013).
“Beginning in the 1990s, it has become increasingly clear that some
autosomal mammalian genes share similarities with the genes that
are subject to X-chromosome inactivation. The defining feature of
these autosomal genes is that, like X-inactivated genes, they are
monoallelically expressed in a random [but stably maintained across
mitotic cell divisions; see previous paragraph] manner. For some genes,
half of the cells express the maternal allele and half of the cells
express the paternal allele; additional genes are monoallelically
expressed in only a subset of cell types but are biallelically
expressed in other cell types. These genes have an ‘all or none’
pattern, such that the non-expressed alleles seem to be completely
or almost completely silent in those cells in which they are not
expressed”. Moreover, this pattern is not a minor one: “New tools ...
are now revealing that there are perhaps more genes that are subject to
random monoallelic expression on mammalian autosomes than there are on
the X chromosome and that these expression properties are achieved by
diverse molecular mechanisms” (Chess 2012).
- Autosomal monoallelic expression was first recognized as happening with immunoglobulin and T cell receptor genes [need a separate section on massive genome remodeling in the immune system, which is coordinated with transcriptional processes], and then with olfactory receptor genes, which account for about 5% of mammalian genes.
- “Autosomal monoallelic expression can have an impact on biological function by affording cells a unique specificity when the products of heterozygous loci might otherwise compete” (Chess 2012).
- Autosomal monoallelic expression “also enhances the phenotypic heterogeneity that is possible in a population of cells” (Chess 2012). “One intriguing possibility is that monoallelic gene expression drives variability between otherwise identical cell types, which are then selected for, either during development or in disease situations” (Eckersley-Maslin and Spector 2014a).
- “Intriguingly, monoallelic expression can result in multiple different outcomes at the transcriptional level. Although there is a general trend for monoallelically expressing cells to have fewer transcript levels than biallelically expressing cells, reduced transcript levels is not a general rule for all monoallelically expressed genes. Indeed, 8% of monoallelically expressed genes in mouse neural progenitor cells show evidence of transcriptional compensation, in that the single active allele in monoallelically expressing cells is upregulated approximately twofold, such that the total transcript levels match that of biallelically expressing cells” (Eckersley-Maslin and Spector 2014a).
- Factors involved in autosomal monoallelic gene expression are thought to include the CTCF protein (see Insulator protein CTCF (CCCTC-binding factor) under THREE-DIMENSIONAL ORGANIZATION OF CHROMOSOMES, NUCLEUS, AND CELL); DNA methylation (see immediately below, and also DNA methylation under PRE-TRANSCRIPTIONAL DECISION-MAKING above); and long noncoding RNAs “which can recruit chromatin modifying factors, and whose deletion or insertion can cause large-scale chromatin reorganization” (Savova, Vigneau and Gimelbrant 2013).
- A different sort of monoallelic expression: “In this study, we uncovered a stochastic pattern of monoallelic expression that differs from the stable allelic regulation of genomic imprinting and allelic exclusion. ... The rapid expression dynamics that we uncovered in individual cells are consistent with models of transcriptional bursting. In each cell, independent bursts of transcription occur from both alleles over time, but RNA from only one allele is often present at any given time. ... It is likely that stochastic transcription of heterozygous alleles contributes to variable expressivity—phenotypic variation among cells and individuals of identical genotypes—which may have fundamental implications for variable disease penetrance and severity” (Deng, Ramsköld, Reinius and Sandberg 2014).
-
Allele-specific DNA methylation. This can result from a DNA
polymorphism on one of two homologous chromosomes, leading to
differential methylation on the two chromosomes and major differences
in expression between two homologous genes or (if the differential
methylation occurs at a regulatory locus) it can lead to changes in the
expression of the various genes regulated by the locus.
(Allele-specific DNA methylation plays a major role in
imprinting.)
- In a study of induced pluripotent cells that were allowed to differentiate into neural progenitor cells: “Our results suggest that random allelic expression imbalance is established during lineage commitment and is associated with increased DNA methylation at the gene promoter”. About 0.65% of the expressed genes showed allelic imbalance in expression (Jeffries, Uwanogho, Cocks et al. 2016, doi:10.1261/rna.058347.116).
-
Allele-specific histone modifications.
These are fairly rare,
but seem to “play an important role in human development. The location
of sites of allele-specific histone modification at key imprinted and
allele-specific expression loci and at sites associated with
developmental disorders suggests that allele-specific histone
modification is an important but as yet undercharacterized phenomenon
involved in embryonic development. (Prendergast, James G. D., Pin Tong,
David C. Hay et al. 2012. This study was restricted to human
embryonic stem cells. Wider investigations remain to be conducted.)
“The monoallelic expression of many imprinted genes in mammals depends on
DNA methylation marks that originate from the germ cells. Recent studies
in mice and fruit flies evoke a novel, transient mode of genomic
imprinting in which oocyte-acquired histone H3 Lys27 trimethylation
(H3K27me3) marks are transmitted to the zygote and modulate the allele
specificity and timing of gene expression in the early embryo”
(Pathak and Feil 2017, doi:10.1038/nsmb.3456).
-
Cis-regulatory polymorphism
“Polymorphisms in cis-regulatory sequences can lead to
differences in levels of expression between the two alleles that
can be extreme (greater than tenfold difference) or can be more
subtle. Even subtle expression differences are still potentially
important as a mechanism that has an impact on genotype-phenotype
correlation” (Chess 2012).
-
Random allelic bias
“This is a lesser-explored mode of gene regulation and refers to
a type of random monoallelic expression wherein some or all
individuals randomly express one of the two alleles preferentially.
Instead of randomness at the cellular (or clonal) level, in random
allelic bias the entire animal or person might have expression
skewed in all cells towards one of the two alleles for a given
gene. Although there are hints of this type of gene regulation in
analyses of allele-specific DNA methylation and mRNA expression,
further consideration of this interesting possible mode of random
monoallelic expression awaits further experimental support” (Chess
2012).
[Parts of this section are misplaced, and belong under “DECISION-MAKING RELATING TO TRANSLATION”.]
Synonymous codons are DNA triplets — sequences of three nucleotide bases,
or “letters” — that code for the same amino acid. Differential usage of
alternative, synonymous codons can affect gene expression, which is to say
that “synonymous” codons have turned out not really to be synonymous: “The
use of particular codons in a genome can increase the expression of a gene
by more than 1000-fold” (Novoa and Pouplana 2012). Likewise, within a
given organism the abundances of tRNAs that recognize particular codons —
and the post-transcriptional modifications of nucleotides within these
tRNAs — are relevant to gene expression (Novoa and de Pouplana (2012).
Much of this is quite new. “Owing to the dogma that the structure (and
therefore function) of proteins is determined by the amino acid sequence,
synonymous mutations were, until recently, referred to as silent” (Sauna
and Kimchi-Sarfaty 2011).
“Codon usage bias, the preference for certain synonymous codons, is found in
all genomes. Although synonymous mutations were previously thought to be
silent, a large body of evidence has demonstrated that codon usage can play
major roles in determining gene expression levels and protein structures.
Codon usage influences translation elongation speed and regulates
translation efficiency and accuracy. Adaptation of codon usage to tRNA
expression determines the proteome landscape. In addition, codon usage
biases result in nonuniform ribosome decoding rates on mRNAs, which in turn
influence the cotranslational protein folding process that is critical for
protein function in diverse biological processes. Conserved genome-wide
correlations have also been found between codon usage and protein
structures. Furthermore, codon usage is a major determinant of mRNA levels
through translation-dependent effects on mRNA decay and
translation-independent effects on transcriptional and posttranscriptional
processes”
(Liu, Yang and Zhao 2021, doi:10.1146/annurev-biochem-071320-112701).
- In one experiment where researchers made synonymous substitutions one-by-one in an mRNA “coding for” green fluorescent protein in the bacterium Escherichia coli, they found the highest-expressing form producing 250 times as much protein as the lowest-expressing form (Kudla et al. 2009).
- Some of the gene-expression differences associated with synonymous codons result from the fact that alternative codons can lead to differently folded RNAs, with important consequences. Folding can affect translation efficiency and mRNA degradation, among many other things. See “RNA structure” under “DECISION-MAKING RELATING TO TRANSLATION” below and also “RNA structure and dynamis” under “OTHER ASPECTS OF THE MOLECULAR STRUCTURE AND DYNAMICS OF DNA AND RNA”.
- mRNA splicing processes can respond differently to distinct but synonymous codons. For example, the synonymous alteration of a codon in an exon (protein-coding segment) of a gene can result in the exon being skipped during splicing (Sauna and Kimchi-Sarfaty 2011). In eukaryotes “a particular bias in codon usage was observed, showing a higher presence of rare codons associated with constraints due to splicing boundaries. Moreover, in some cases, a particular codon bias was related to regulatory enhancers of splicing elements in several genes such as the tumor suppressor TP53” (Marin 2008).
- Synonymous codons can differentially affect mRNA stability. In the case of reduced global stability, lower protein levels may result. And greater local stability near the start codon may impede translation initiation and therefore also lower the protein levels (Sauna and Kimchi-Sarfaty 2011).
- In a group of synonymous codons, some will be translated by the ribosome faster than others. If faster codons tended to occur earlier in the translation process (toward the beginning of the mRNA), then successive ribosomes processing the mRNA would speed through the early parts and then, upon reaching the slower codons, back up against each other, probably resulting in incomplete translation and toxic proteins when some ribosomes disengaged from the mRNA. In a study of highly expressed mRNAs (that is, those most likely to have multiple engaged ribosomes), it was found that these mRNAs are indeed “front-loaded” with slower codons to prevent this problem.
- “The sets of genes that are expressed in each stage of the cell cycle present similar codon covariations, and these differ from those found in other stages, suggesting that the codon preferences change during the cell cycle” (Novoa and de Pouplana 2013).
- Another regulatory element: Like nearly everything else, tRNAs are not fixed, unchangeable elements, but are themselves subject to modification by enzymes that “alter their translation decoding capacity, potentially imipacting the subset of ‘preferred’ codons in the genome. This potential variability in the sets of ‘preferred’ codons implies that modulating the activity of modification enzymes may be an avenue for regulating the composition of the proteome when needed”. Over 100 post-transcriptional modifications of tRNA nucleotides have been described, and they “contribute to tRNA folding, structure, and stability, as well as to translation efficiency and amino acid substitution rates. ... Increasing evidence indicates that tRNA modifications can have regulatory roles in cells, especially in response to stress conditions”. “Similar transcriptomes may result in different proteome compositions as a consequence of changes in the activity of anticodon modification enzymes” (Novoa and de Pouplana 2013).
- It’s been found in Escherichia coli that, whereas proteins enriched for different synonymous codons have overall translation rates that do not greatly differ under normal conditions, the rates can differ up to a hundred-fold in stressed environments, where particular amino acids are in reduced supply. This enables production of some proteins to continue under the stressful conditions, while synthesis of whole classes of other proteins is more or less shut down (Subramaniam, Pan and Cluzel: advance epub 2012).
- Synonymous codons can also result in differently folded proteins, and therefore in proteins with different functions. This is thought to be because the choice of codon can affect the speed of translation, which in turn affects the co-translational folding process (as well as protein abundance). “A single protein that is prone to misfolding can lead to a cascade effect of misfolding in other proteins and, eventually, to proteotoxicity” (Sauna and Kimchi-Sarfaty 2011).
- Likewise, synonymous codons on an mRNA may generate translation “pause sites”, which influence how the resulting protein folds (Sauna and Kimchi-Sarfaty 2011). Putting this together with the significance of translation speed, one reviewer of the literature offered this comparison: “Making an analogy with musical language, there is some similarity to syncopation: the execution of fragments with the same notes, same compass, can produce a completely different effect due to unexpected changes of rhythm” (Marin 2008).
- There are direct functional consequences for this kind of thing. The replacement of 16 consecutive rare codons with frequent codons in a particular enzyme “led to a 20-30% reduction in the enzymatic activity [of the enzyme] ... In addition, the profile of translation pauses due to the synonymous codon change was modified. Thus, the substitution of 16 rare codons modified the translation kinetics, reducing the biological activity of the protein. More recently, the substitution of a single codon proved to be sufficient to modify translation pausing” (Marin 2008).
- Not only folding of a protein, but even the post-translational modifications of that protein may be affected by synonymous codons. Higher vertebrates produce actin via six copies of a gene that are nearly identical at the amino acid-coding level, but whose functions in the cell are only minimally redundant. The key is that mRNAs for these proteins are substantially different due to differential use of synonymous codons. By affecting translation rate and protein folding, the different mRNAs lead to different post-translational modifications (which are in this case applied during the translation process), with consequent differences in protein function (Shabalina, Spiridonov and Kashina 2013).
- Synonymous codons can differentially affect nucleosome positioning, with all the implications discussed under “Nucleosome positioning”, above. (See Wilke and Drummond 2010 for literature references.)
- Synonymous codons in protein-coding genes can be under evolutionary constraint, and “further analysis suggests that these sites affect RNA-transcript processing, microRNA binding and how chromatin states are established” (Baker 2012). For example, a gene specifying a protein involved in removing intracellular bacteria may mutate by substituting a synonymous codon at a microRNA binding site. With the microRNA now blocked, the gene gets over-expressed, and happens to have the effect of inhibiting the anti-bacterial activity and thereby worsening an illness (Katsnelson 2011).
- “A number of high-profile studies have suggested that synonymous mutations may indeed play a causal role in cancer progression” (Hofree, Shen, Carter et al. 2013a; see this article for references). More generally: “Upwards of 50 disorders — including depression, schizophrenia, multiple cancers, cystic fibrosis and Crohn’s disease — have now been linked to synonymous mutations. ... In one recent inspection of more than 2,000 human genome studies, for example, a team from Stanford University School of Medicine in California found that synonymous mutations were just as likely as nonsynonymous ones to play a part in disease mechanisms”. “At the moment”, according to Laurence Hurst at the University of Bath in the UK, “we are discovering the major mechanisms by which synonymous mutations can be associated with disease. And they are vastly more diverse than people thought” (Katsnelson 2013).
- “We report that synonymous codon choice is tuned to promote interaction of nascent polypeptides with the signal recognition particle (SRP), which assists in protein translocation across membranes. Cotranslational recognition by the SRP in vivo is enhanced when mRNAs contain nonoptimal codon clusters 35–40 codons downstream of the SRP-binding site, the distance that spans the ribosomal polypeptide exit tunnel. A local translation slowdown upon ribosomal exit of SRP-binding elements in mRNAs containing these nonoptimal codon clusters is supported experimentally by ribosome profiling analyses in yeast. Modulation of local elongation rates through codon choice appears to kinetically enhance recognition by ribosome-associated factors”. One result of all this is delivery of the newly generated protein to particular membrane sites. The authors propose that codon choices affecting the rate of translation by the ribosome may be a general feature of translation (and therefore of gene expression regulation). (doi:10.1038/nsmb.2919)
- In Drosophila: “we showed that [in the circadian protein] dper codon usage is important for circadian clock function. Codon optimization of dper resulted in conformational changes of the dPER protein, altered dPER phosphorylation profile and stability, and impaired dPER function in the circadian negative feedback loop, which manifests into changes in molecular rhythmicity and abnormal circadian behavioral output ... These results suggest a universal mechanism in eukaryotes that uses a codon usage ‘code’ within genetic codons to regulate cotranslational protein folding” (Fu, Murphy, Zhou et al. 2016, 10.1101/gad.281030.116).
- In sum, “As we begin to decipher some of the rules that govern codon usage and tRNA abundances, it is becoming clear that these parameters are a way to not only increase gene expression, but also regulate the speed of ribosomal translation, the efficiency of protein folding, and the coordinated expression of functionally related gene families” (Novoa and Pouplana 2012).
The DNA in mitochondria (comprising 37 genes in humans) is often scarcely
an afterthought in genetic studies, but now is gaining increasing
importance.
Viral DNA present in cells is also beginning to figure in genetic studies.
Tens of thousands of short, extrachromosomal, circular DNAs (microDNAs)
have been found in mouse and human tissues. They are 200 — 400 base pairs
long and correspond to deletions from genic portions of DNA in affected
cells. “The results suggest that microdeletions occur in an average of 1
in 2000 chromosomal DNA molecules” (Shibata, Kumar, Layer et al. 2012).
In other words, something like 1 in 44 cells would exhibit such deletions.
-
Mitochondrial and viral DNA
On mitochondria and gene expression generally: “The nuclear epigenome
modulates mitochondrial function in numerous ways. Conversely, metabolic
signals originating from the mitochondria can initiate epigenetic
modifications in the nucleus. This reciprocal relationship between
mitochondria and the nuclear genome is a multilayered process that also
involves an important epigenetic component. We are only beginning to
understand the full complexity of this regulation”. “Crosstalk between
mitochondria and the nuclear epigenome represents bidirectional
mitonuclear communication: mitochondria are essential mediators of
epigenetic processes [for example, histone acetylation and histone and
DNA methylation] and, conversely, changes in epigenome regulate
mitochondrial function”. “Mitochondria-mediated changes in the epigenome
affect stress responses and longevity” (Levis and Pfennig 2016,
doi:10.1016/j.tree.2016.03.012).
- Researchers studying the effects of the presence versus the absence of various genes in yeast were puzzled to find that their results couldn’t be reliably duplicated. Eventually they discovered that the differing mitochondria in the yeast lines they were using, and the presence of a virus, had a dramatic effect on the phenotypes relating to the genes they were studying. “We have shown that both the source of the mitochondrial genome and the presence or absence of a double-stranded DNA virus influence the phenotype of chromosomal variants that affect the growth of yeast”. The researchers expect these results to apply to humans once appropriate studies are conducted. They note that “recent work on a mouse model of Crohn disease supports a combinatorial model of complex disease traits in which the pathology requires the interaction between a specific mutation in the mouse and a specific strain of virus” (Edwards, Symbor-Nagrabska, Dollard et al. 2014).
- Speaking of mitochondrial and viral DNA: “Nonchromosomal information is not under the usual constraints of the nuclear genome. These nonchromosomal elements are extremely unstable: they mutate at higher frequencies than the DNA of the chromosomal genome, may be lost at high frequencies without loss of viability, and can vary in copy number from cell to cell” (Edwards, Symbor-Nagrabska, Dollard et al. 2014).
-
DNA microdeletions and circular DNA
- “Chromosomal loci that are enriched sources of microDNA in the adult brain are somatically mosaic for microdeletions that appear to arise from the excision of micro DNAs”. (Shibata, Kumar, Layer et al. 2012).
- “The generation of microDNAs and microdeletions may produce a large pool of individual-specific or somatic-clone–specific copy-number variations of small segments of the genome. The genetic mosaicism in somatic tissues may lead to functional differences between cells in a tissue. Finally, persistent microDNAs may provide the extrachromosomal genetic ‘cache’ that has been postulated to account for non-Mendelian genetics in plants” (Shibata, Kumar, Layer et al. 2012).
- Evidence suggests that the same sort of microdeletion occurs in germline cells (Shibata, Kumar, Layer et al. 2012).
- See also “Extracellular genomic DNA fragments” under MISCELLANEOUS below.
- In addition to their post-transcriptional role (see “Small interfering RNAs (siRNAs)” under NONCODING RNA below), siRNAs act directly on chromatin, playing a role, for example, in DNA methylation.
MicroRNAs can target gene coding regions and perhaps also promoters,
thereby reducing gene expression. See
“microRNA (miRNA) activity” under
NONCODING RNA below.
“Many metabolites have been shown to have a direct effect on gene
expression patterns through binding to nuclear receptors that in turn
affect the transcription of the gene they bind to. Interestingly, even
transient changes in the nutrition can have a long-lasting impact on gene
expression patterns. This memory of former metabolic states may also be
involved in disease progression” (Katada, Imhof and Sassone-Corsi 2012).
“A prominent area in epigenetic research that has emerged in recent years
relates to how cellular metabolism regulates various events of chromatin
remodeling. Cells sense changes in the environment and translate them into
specific modulations of the epigenome through a variety of signaling
components, several of which are proteins with histone- and DNA-modifying
enzymatic activity. There are now a myriad of residues on DNA and histone
tails that can undergo modification at a given time. The enzymes that elicit
these modifications rely critically on the availability of phosphate,
acetyl, and methyl groups, to mention a few. This constitutes an intriguing
link between cellular metabolism and epigenetic control that has previously
been largely unappreciated ... a number of remarkable studies discussed in
this article are revealing a range of responses to the environment”
(Berger and Sassone-Corsi 2016, doi:10.1101/cshperspect.a019463).
“Chromatin regulation involves enzymes that use cofactors for the reactions
that modify DNA or histones. These enzymes either attach small chemical
units (i.e., posttranslational modifications or PTMs) or alter nucleosome
positioning or composition (i.e., of histone variants). It is assumed that
this control depends partly on the variable levels of cellular metabolites
acting as enzyme cofactors. For example, acetyltransferases use
acetyl-coenzyme A (acetyl-CoA), methyltransferases use S-adenosyl
methionine, and kinases use ATP as donors of acetyl, methyl, or phospho
groups, respectively; deacetylases can use nicotinamide adenine dinucleotide
(NAD), and demethylases can use flavin adenine dinucleotide (FAD) or
α-ketoglutarate as coenzymes. In addition, another relevant example relates
to remodeler complexes that use ATP for moving, ejecting, or restructuring
nucleosomes”
(Berger and Sassone-Corsi 2016, doi:10.1101/cshperspect.a019463).
“One key proposal to come from the studies of histone acetyltransferases and
histone deacetylases is that many other epigenetic enzymes may also nimbly
interact with the environment via their response to changing concentrations
of metabolites. The ‘circadian epigenome’ and the ‘aging epigenome’
represent examples of striking physiological states that are influenced by
metabolic changes, which impinge on chromatin. Many more physiological
states are altered by metabolic epigenetics, such as numerous types of
cancers; many others await elucidation”
(Berger and Sassone-Corsi 2016, doi:10.1101/cshperspect.a019463).
“Metabolism and gene expression, which are two fundamental biological
processes that are essential to all living organisms, reciprocally regulate
each other to maintain homeostasis and regulate cell growth, survival and
differentiation. Metabolism feeds into the regulation of gene expression via
metabolic enzymes and metabolites, which can modulate chromatin directly or
indirectly — through regulation of the activity of chromatin trans-acting
proteins, including histone-modifying enzymes, chromatin-remodelling
complexes and transcription regulators. Deregulation of these metabolic
activities has been implicated in human diseases, prominently including
cancer.” (Li, Egervari, Wang et al. 2018, doi:10.1038/s41580-018-0029-7)
“Susceptibility of the epigenome to altered metabolic cofactor availability
provides a direct path for metabolism to regulate the transcriptional
environment of cells and organisms. This allows chromatin to act as a signal
integrator of diverse metabolic pathways, like those described here, to
regulate both canonical (e.g., acetylation and methylation) and emerging
(e.g., acylation, ADP-ribosylation, O-GlcNAcylation, and lactylation)
epigenetic modifications. Numerous studies have provided evidence in
support of this general model, yet it remains largely unclear why certain
regions of the epigenome are more or less susceptible to metabolic
perturbations than others. Recent insights into the capabilities of
loci-specific regulation of the epigenome by metabolic enzymes introduces
potential mechanisms for more directed regulation of chromatin modifications
during fluctuations in cofactor availability. However, these metabolic
enzymes cannot act in isolation as the PTMs they support require coordinated
regulation of relevant chromatin effectors to facilitate a functional
transcriptional response. Broad experimental approaches (i.e., biochemical,
genetic, physiological, etc.) will be needed to uncover such complex
mechanisms as the metabolism-epigenome relationship
paradigm continues to shift”
(Haws, Leech, and Denu 2020, doi:10.1016/j.tibs.2020.04.002).
- “A new mechanistic link between metabolic flux and regulation of gene expression is through moonlighting of metabolic enzymes in the nucleus. This facilitates delivery of membrane-impermeable or unstable metabolites to the nucleus, including key substrates for epigenetic mechanisms such as acetyl-CoA which is used in histone acetylation. This metabolism–epigenetics axis facilitates adaptation to a changing environment in normal (e.g., development, stem cell differentiation) and disease states (e.g., cancer) ... Many cytoplasmic metabolic enzymes (including all essential glycolytic enzymes) and mitochondrial enzymes moonlight in the nucleus ... These nuclear metabolic enzymes provide the basis of an emerging metabolism-gene transcription axis, which includes epigenetic regulation (histone acetylation, histone and DNA methylation). Growing evidence suggests that this axis optimizes adaptive responses linking metabolic stress to cellular functions such as proliferation or differentiation” (Boukouris, Zervopoulos and Michelakis 2016, doi:10.1016/j.tibs.2016.05.013).
- Small peptides (smaller than the lower limit of 100 or so amino acids usually assumed in scans for proteins) have been found to excise part of a transcription factor, thereby changing the factor from a repressor of gene action to an activator and playing a role in regulating gene expression during development. (The work was done in fruit flies.) The peptides derive from a long noncoding RNA, segments of which encode tiny peptides (Rosenberg and Desplan 2010, reporting on work by T. Kondo et al.).
- “We analyze the translatomes of 80 human hearts to identify new translation events and quantify the effect of translational regulation ... We identify hundreds of previously undetected microproteins, expressed from lncRNAs and circRNAs, for which we validate the protein products in vivo. The translation of microproteins is not restricted to the heart and prominent in the translatomes of human kidney and liver. We associate these microproteins with diverse cellular processes and compartments and find that many locate to the mitochondria. Importantly, dozens of microproteins are translated from lncRNAs with well-characterized noncoding functions, indicating previously unrecognized biology” (van Heesch, Witte, Schneider-Lunitz et al. 2019, doi:10.1016/j.cell.2019.05.010).
- Various heavy metal ions can result in deformation of the DNA double helix, a shift in the position of DNA on nucleosomal histones, alteration of histone modifications, and (in the case of nickel) hypermethylation of DNA — all with gene regulation implications. These ions have mostly been studied in relation to gene dysregulation in carcinogenesis, but they presumably also function in healthy states (Mohideen, Muhammad and Davey 2010).
- It appears that so-called “hyperedited” double-stranded RNAs — themselves the result of post-transcriptional regulation of gene expression via RNA editing (see below) — are in turn involved in regulation of gene transcription. The presence of such RNAs inhibits expression of certain genes involved in immunity and defense by blocking the stimulation of those genes by interferon (Vitali and Scadden 2010).
-
RNA polymerase
RNA polymerase does not merely transcribe genes mechanically; it is a major
regulator of gene expression.
“The large subunit of Pol II contains an intrinsically disordered C-terminal
domain that is phosphorylated by cyclin-dependent kinases during the
transition from initiation to elongation, thus influencing the interaction
of the C-terminal domain with different components of the initiation or the
RNA-splicing apparatus ... Both the transcription-initiation machinery and
the splicing machinery can form phase-separated condensates that contain
large numbers of component molecules: hundreds of molecules of Pol II and
mediator are concentrated in condensates at super-enhancers and large
numbers of splicing factors are concentrated in nuclear speckles, some of
which occur at highly active transcription sites ... We find that the
hypophosphorylated C-terminal domain of Pol II is incorporated into mediator
condensates and that phosphorylation by regulatory cyclin-dependent kinases
reduces this incorporation. We also find that the hyperphosphorylated
C-terminal domain is preferentially incorporated into condensates that are
formed by splicing factors. These results suggest that phosphorylation of
the Pol II C-terminal domain drives an exchange from condensates that are
involved in transcription initiation to those that are involved in RNA
processing, and implicates phosphorylation as a mechanism that regulates
condensate preference”
(Guo, Manteiga, Henninger et al. 2019, doi:10.1038/s41586-019-1464-0).
“Microscopy studies have revealed that transcription involves the
condensation of factors in the cell nucleus. A model is emerging for the
transcription of protein-coding genes in which distinct transient
condensates form at gene promoters and in gene bodies to concentrate the
factors required for transcription initiation and elongation, respectively.
The transcribing enzyme RNA polymerase II may shuttle between these
condensates in a phosphorylation-dependent manner”
(Cramer 2019, doi:10.1038/s41586-019-1517-4).
See also “Phase transitions and membraneless
organelles” below.
Two studies “show how ubiquitylation of a single lysine residue in RNA
polymerase II serves as a master switch to regulate transcription, RNA
polymerase II degradation, and transcription-coupled nucleotide excision
repair in response to DNA damage”.
(Son and Schärer 2020, doi:10.1016/j.cell.2020.02.053).
“Existing models suggest that RNA polymerases I and III (Pol I and Pol
III) are the only enzymes that directly mediate the expression of the
ribosomal RNA (rRNA) components of ribosomes. Here we show, however, that
RNA polymerase II (Pol II) inside human nucleoli operates near genes
encoding rRNAs to drive their expression. Pol II, assisted by the
neurodegeneration-associated enzyme senataxin, generates a shield comprising
triplex nucleic acid structures known as R-loops at intergenic spacers
flanking nucleolar rRNA genes. The shield prevents Pol I from producing
sense intergenic noncoding RNAs (sincRNAs) that can disrupt nucleolar
organization and rRNA expression. These disruptive sincRNAs can be unleashed
by Pol II inhibition, senataxin loss, Ewing sarcoma or locus-associated
R-loop repression through an experimental system”
(Abraham, Khosraviani, Chan et al. 2020, doi:10.1038/s41586-020-2497-0).
- “RNA polymerase III (Pol III) is tightly controlled in response to environmental cues ... Here, we describe genome-wide studies in human fibroblasts that reveal a dynamic and gene-specific adaptation of Pol III recruitment to extracellular signals in an mTORC1-dependent manner. Repression of Pol III recruitment and transcription are tightly linked to MAF1, which selectively localizes at Pol III loci ... and increasingly targets transcribing Pol III in response to serum starvation ... We show that Pol III occupancy closely reflects ongoing transcription. Our results ... identify previously uncharacterized, differential coordination in Pol III binding and transcription under different growth conditions” (Orioli, Praz, Lhôte and Hernandez 2016, 10.1101/gr.201400.115).
- “RNA polymerase II (Pol II) transcription termination by the Nrd1p-Nab3p-Sen1p (NNS) pathway is critical for the production of stable noncoding RNAs and the control of pervasive transcription in Saccharomyces cerevisiae ... We found that nucleosomes and specific DNA-binding proteins, including the general regulatory factors (GRFs) Reb1p, Rap1p, and Abf1p, and Pol III transcription factors enhance the efficiency of NNS termination by physically blocking Pol II progression ... Reduced binding of these factors results in defective NNS termination and Pol II readthrough. Furthermore, inactivating NNS enables Pol II elongation through these roadblocks, demonstrating that effective Pol II termination depends on a synergy between the NNS machinery and obstacles in chromatin” (Roy, Gabunilas, Gillespie et al. 2016, doi:10.1101/gr.204776.116).
-
One of countless molecules bearing on Pol II transcription:
“The conserved, multifunctional Polymerase-Associated Factor 1 complex
(Paf1C) regulates all stages of the RNA polymerase (Pol) II transcription
cycle. [Recent studies] identify new roles for Paf1C in the control of
gene expression and the regulation of chromatin structure. In exploring
these advances, we find that various functions of Paf1C, such as the
regulation of promoter-proximal pausing and development in higher
eukaryotes, are complex and context dependent”. In particular:
- “Paf1C can be recruited to genes by transcriptional activators and through interactions with the Pol II elongation machinery.”
- “Paf1C can function to maintain promoter-proximal pausing of Pol II or to promote release from pausing, depending upon the genetic context.”
- “Paf1C regulates cleavage and polyadenylation of mRNA, governs polyA site selection, and controls the export of nascent transcripts.”
- “Paf1C controls chromatin structure by promoting several cotranscriptional histone modifications and is important for the establishment of proper boundaries between heterochromatin and euchromatin.”
- “Paf1C regulates pluripotency and development in higher eukaryotes, and several new studies link Paf1C misregulation to cancer.”
-
RNA polymerase pausing, release, and elongation
RNA polymerase II does not simply initiate transcription of a
protein-coding gene and then proceed to completion, releasing the mRNA
it has produced. There are several steps, each subject to elaborate
regulation: (1) Formation of the pre-initiation complex (PIC) as
described under “Pre-initiation complex” above. (2)
After some 20 – 60 nucleotides have been transcribed, RNA pol II —
still not completely clear of the influence of the gene promoter and
pre-initiation complex — is held back (paused) by various factors.
“The duration of pausing depends on the rate of recruitment of factors
that trigger pause release, which is variable from gene to gene and
under different cell conditions” (Fromm, Gilchrist and Adelman 2013).
(3) An enzyme subject to recruitment by various methods (themselves
subject to regulation) may eventually phosphorylate the factors
associated with RNA pol II and responsible for pausing, resulting in
“pause release”. (4) Productive elongation occurs, with all the
regulatory processes associated with nucleosomes and histone
modifications (discussed under various headings above) potentially
coming into play.
“RNA polymerase II (Pol II) assembles with basal transcription factors
into the transcription pre-initiation complex (PIC) on active promoters.
Following transcription initiation, Pol II pauses 30-50 bp downstream of
the transcription start site (TSS), and requires further activation to
proceed to productive transcription elongation. Some promoters have a
greater tendency for Pol II pausing; these promoters better mediate
transcriptional responses to developmental or environmental cues ... In
summary, Pol II pausing can be persistent and can inhibit transcription
reinitiation. This can be associated with genes that have lower
steady-state expression levels, such as those that are responsive to
external cues. Thus, Pol II pausing at these genes could prevent
transcription in the absence of stimuli while simultaneously maintaining
the promoters in a poised state for signal-induced activation”
(Zlotorynski 2017, doi:10.1038/nrm.2017.57).
“Accumulation of Pol II near most promoters demonstrates that relative
rates of termination and pause release are much slower than rates of
recruitment and initiation. Regulatory processes typically target
rate-limiting steps, and, consistently, the release of paused Pol II has
emerged as a central point of gene control ... there is growing evidence
for rapid Pol II turnover at certain metazoan genes and enhancers,
suggesting an underappreciated layer of transcriptional control”. “Potent
small molecule inhibitors that block pause release were demonstrated to
broadly trap Pol II at promoters and abrogate nearly all RNA synthesis in
Drosophila and mammalian cells”
(Core and Adelman 2019, doi:10.1101/gad.325142.119).
In sum, “key steps regulating transcription occur after Pol II has
associated with a gene’s promoter” (Li and Gilmour 2011). “Genome-wide
data in metazoans now point to the widespread importance of Pol II
pausing in transcription regulation. Indeed, the escape of paused Pol
II into productive elongation is regulated during environmental stress,
immunological signalling and development” (Adelman and Lis 2012).
“We show that RNAPII pauses at PARP1–chromatin structures within the gene
body. Knockdown of PARP1 abolishes this RNAPII pausing, suggesting that
PARP1 may regulate RNAPII elongation. Additionally, PARP1 alters
nucleosome deposition and histone post-translational modifications at
specific exon–intron boundaries, thereby affecting RNAPII movement.
Lastly, genome-wide analyses confirmed that PARP1 influences changes in
RNAPII elongation by either reducing or increasing the rate of RNAPII
elongation depending on the chromatin context.”
(Matveeva, Al-Tinawi, Rouchka et al. 2019, doi:10.1186/s13072-019-0261-1)
“The high degree of conservation in protein sequences thought to be
unstructured has hinted that these regions may have important biological
functions. Although unstructured regions are widely viewed to be crucial
for protein signaling, localization, and stability, their roles in many
other settings have remained mysterious. Cermakova et al. discovered that
prominent members of the transcription elongation machinery are linked
through a network of interactions involving transcription elongation
factor TFIIS N-terminal domains (TNDs) and conserved unstructured
sequences called ‘TND-interacting motifs’ (TIMs). The researchers found
that mutation of a single TIM in a central organizing protein of this
network abolished key protein interactions and induced widespread defects
in transcription elongation dynamics”
(Cermakova, Demeulemeester, Lux et al. 2021, doi:10.1126/science.abe2913).
“Trimethylation of histone H3 lysine 4 (H3K4me3) is associated with
transcriptional start sites and has been proposed to regulate
transcription initiation ... Acute loss of H3K4me3 does not have
detectable effects on transcriptional initiation but leads to a
widespread decrease in transcriptional output, an increase in RNA
polymerase II (RNAPII) pausing and slower elongation. We show that
H3K4me3 is required for the recruitment of the integrator complex subunit
11 (INTS11), which is essential for the eviction of paused RNAPII and
transcriptional elongation. Thus, our study demonstrates a distinct role
for H3K4me3 in transcriptional pause-release and elongation rather than
transcriptional initiation”
(Wang, Fan, Shliaha et al. 2023, doi:10.1038/s41586-023-05780-8).
“How enhancers control target gene expression over long genomic distances
remains an important unsolved problem. [Experiments show] that enhancers
spend more time in close proximity to their target promoters in
functional enhancer–promoter pairs compared to nonfunctional pairs, which
can be attributed in part to factors unrelated to genomic position.
Manipulation of the transcription cycle demonstrated a key role for Pol
II in enhancer–promoter interactions. Notably, promoter-proximal paused
Pol II itself partially stabilized interactions. We propose an updated
model in which elements of transcriptional dynamics shape the duration or
frequency of interactions to facilitate enhancer–promoter communication”
(Barshad, Lewis, Chivu et al. 2023, doi:10.1038/s41588-023-01442-7).
For a convenient (if now somewhat dated) overview, see “SnapShot:
Transcription Regulation: Pausing” by Fromm, Gilchrist and Adelman in the
May 9, 2013 issue of Cell.
- “>55% of non-expressed genes in mouse embryonic stem cells have an accumulation of RNA polymerase II at their promoter”. “This is either a regulated blockage of transcription until a release or activation signal is received (referred to as ‘poising’), or it is an accumulation of RNAPII at the promoter of actively transcribed genes that is due to RNAPII slowing down immediately downstream of the TSS (most often referred to as ‘pausing’)” (Lenhard, Sandelin and Carninci 2012).
- “Recent findings indicate that progression of a promoter-proximal, paused RNAPolII [RNA polymerase II] into productive elongation is a rate limiting step in the transcription of nearly 40% of genes in mouse embryonic stem cells and mouse embryonic fibroblasts. Interestingly, key pluripotency regulatory genes...exhibit a regulated rate of escape from pausing, suggesting that RNApolII pausing may provide a responsive transcriptional regulation control during cell differentiation. At each promoter, a particular combination of transcription factors, elongation factors, nucleosomes and underlying DNA sequence act together to determine the kinetics of the RNApolII capture and release, thus orchestrating the regulation of DNA transcription by this enzyme” (Sequeira-Mendes and Gómez 2011).
- “Pausing intensity and position depend on interactions of the core promoter complex with Pol II and on the first nucleosome barrier, both of which appear to contribute to differing extents on different promoters” (Kwak and Lis 20130).
- Pausing occurs, not only at promoters, but also during elongation, with implications for chromatin remodeling: “A recent study shows that RNAP II pauses frequently throughout the body of genes and each pause occurs just before a nucleosome. In addition to a plethora of transcription activators and chromatin remodeling factors, it appears that RNAP II itself is required to break DNA–histone contacts, at least at the promoter. However, during transcription elongation, RNAP II assumes an even more prominent role in chromatin remodeling. The transition from transcription initiation to elongation depends on phosphorylation of the RNAP II CTD [carboxy-terminal domain], first at Ser5 residues and then at Ser2. This phosphorylation of the CTD creates binding sites for proteins that will modify the histones” (de Almeida and Carmo-Fonseca 2012).
- There are proteins that encourage pausing by inhibiting elongation, and at least one protein that reverses this inhibition. “Cells appear to use...pausing in different ways to either positively or negatively regulate gene expression” (Li and Gilmour 2011). As one example: there is “a functional link between chromatin-associated Hsp90 [heat shock protein 90] and pausing of pol II. We find that Hsp90 preferentially binds transcription start sites that exhibit pol II pausing. Hsp90 controls expression of these target genes by stabilizing NELF [negative elongation factor] complex and thus regulating paused pol II at these loci” (Sawarkar, Seivers and Paro 2012). Hsp90 is responsive to environmental stimuli, and this regulation links such stimuli to gene expression.
- “Pausing during transcription elongation is a fundamental activity in all kingdoms of life. In bacteria, the essential protein NusA modulates transcriptional pausing, but its mechanism of action has remained enigmatic. By combining structural and functional studies we show that a helical rearrangement induced in NusA upon interaction with RNA polymerase is the key to its modulatory function. This conformational change leads to an allosteric re-positioning of conserved basic residues that could enable their interaction with an RNA pause hairpin that forms in the exit channel of the polymerase. This weak interaction would stabilize the paused complex and increases the duration of the transcriptional pause. Allosteric spatial re-positioning of regulatory elements may represent a general approach used across all taxa for modulation of transcription and protein–RNA interactions” (Ma, Mobli, Yang et al. 2015; doi:10.1093/nar/gkv108).
- It appears that pausing can be “viewed as a mechanism to fine-tune gene expression, and to potentiate genes for further or future activation”. A paused RNA polymerase prevents the promoter region from being occupied by nucleosomes, with their frequently repressive effect upon transcription. It also makes possible extremely rapid activation of transcription when circumstances call for it. (Gilchrist, Santos, Fargo et al. 2010)
- In Drosophila “paused Pol II is much more prevalent at genes encoding components and regulators of signal transduction cascades than at inducible downstream targets. Within immune-responsive pathways, we found that pausing maintains basal expression of critical network hubs...We conclude that the role of pausing goes well beyond poising inducible genes for activation and propose that the primary function of paused Pol II is to establish basal activity of signal-responsive networks” (Gilchrist, Fromm, dos Santos et al. 2012).
- “Promoter proximal pausing is not an absolute requirement for either rapid or high induction of gene expression, but appears to be a common feature at genes that are normally expressed at some basal level, but which have the capacity to be rapidly induced by changes in cellular environment. Expression of such genes requires very precise control as too little expression may render the cells unable to respond to incoming signals, and too much may trigger expression of downstream effectors in the absence of the appropriate signal. ... Animal studies confirm that correct regulation of promoter proximal pausing is critical for development and health in adult life” (Jennings 2013).
- In fruit flies, RNA polymerase II pausing has been shown to be crucial for proper synchronous gene expression during early development. For example, the snail gene plays an important role in the coordinated invagination of about 1000 mesoderm cells during gastrulation. Replacement of the promoter for this gene with one that prevents or weakens pausing resulted in severe gastrulation defects. (Research summarized in Burgess 2013.)
- “From genome-wide Pol II occupancy data, the authors [of a particular study] noticed that Pol II occupancy varied in a continuous manner among different genes and hence that previous designations of genes as ‘paused’ or ‘non-paused’ might be oversimplistic. Indeed, using six of these promoters to drive a reporter gene, they showed gradations in the degree of Pol II pausing that correlated with the levels and synchronicity of transcription”. (Research summarized in Burgess 2013.)
- Longer introns in a gene “can increase times between pulses [of transcription], adding yet another checkpoint to the regulation of gene expression" (Papantonis and Cook 2010).
- “Elongation rate may be different between genes and cell types and can both affect and be affected by transcription level and cotranscriptional processing” (Kwak and Lis 2013).
- “Recently, a new multiprotein complex, termed Integrator, has been shown to regulate elongation by recruiting the SEC [super elongation complex]”. (Vernimmen and Bickmore 2015, doi:10.1016/j.tig.2015.10.004)
- “The association of DSIF and NELF with initiated RNA Polymerase II (Pol II) is the general mechanism for inducing promoter-proximal pausing of Pol II ... We show that the release of the paused Pol II is cooperatively regulated by multiple P-TEFbs which are recruited by bromodomain-containing protein Brd4 and super elongation complex (SEC) via different recruitment mechanisms. Upon stimulation, Brd4 recruits P-TEFb to Spt5/DSIF via a recruitment pathway consisting of Med1, Med23 and Tat-SF1, whereas SEC recruits P-TEFb to NELF-A and NELF-E via Paf1c and Med26, respectively. P-TEFb-mediated phosphorylation of Spt5, NELF-A and NELF-E results in the dissociation of NELF from Pol II, thereby transiting transcription from pausing to elongation. Additionally, we demonstrate that P-TEFb-mediated Ser2 phosphorylation of Pol II is dispensable for pause release. Therefore, our studies reveal a co-regulatory mechanism of Brd4 and SEC in modulating the transcriptional pause release by recruiting multiple P-TEFbs via a Mediator- and Paf1c-coordinated recruitment network” (Lu, Zhu, Li et al. 2016, doi:10.1093/nar/gkw571).
- “RNA polymerase II (RNAPII) transcribes chromosomal DNA that contains multiple nucleosomes. The nucleosome forms transcriptional barriers, and nucleosomal transcription requires several additional factors in vivo. We demonstrate that the transcription elongation factors Elf1 and Spt4/5 cooperatively lower the barriers and increase the RNAPII processivity in the nucleosome. The cryo–electron microscopy structures of the nucleosome-transcribing RNAPII elongation complexes (ECs) reveal that Elf1 and Spt4/5 reshape the EC downstream edge and intervene between RNAPII and the nucleosome. They facilitate RNAPII progression through superhelical location SHL(–1) by adjusting the nucleosome in favor of the forward progression. They suppress pausing at SHL(–5) by preventing the stable RNAPII-nucleosome interaction. Thus, the EC overcomes the nucleosomal barriers while providing a platform for various chromatin functions” (Ehara, Kujirai, Fujino et al. 2019, doi:10.1126/science.aav8912).
-
It can be useful to see how little is captured by notes such as the
foregoing — and how little is actually known about all the relevant
processes. What follows is a list of the factors that could be
identified in 2013 as participating in transcriptional elongation
(copied from Kwak and Lis 2013). It’s important to realize that
each of the factors listed here enters the picture out of its own
world of “regulation”. At the molecular level of the organism we
are always looking at ever-widening circles of interaction,
without limit. It’s just a question of how narrowly we choose to
focus our attention — and how much of the context we consequently
block from view.
Class Factor name Function Related factors and notes GAGA factor GAF Generates nucleosome-free region and promoter structure for pausing NURF General Transcription Factors TFIID Generates promoter structure for pausing TFIIF Increases elongation rate Near promoters TFIIS Rescues backtracked Pol II Pol III Pausing factors NELF Stabilizes Pol II pausing DSIF Stabilizes Pol II pausing and facilitates elongation Positive elongation factor P-TEFb Phosphorylates NELF, DSIF, and Pol II CTD for pause release Processivity factors Elongin Increases elongation rate ELL Increases elongation rate AFF4 SEC Contains P-TEFb and ELL Mediator, PAF Activator c-Myc Directly recruits P-TEFb NF-κB Directly recruits P-TEFb Coactivator BRD4 Recruits P-TEFb Mediator Recruits P-TEFb via SEC Capping machinery CE Facilitates P-TEFb recruitment, counters NELF/DSIF RNMT Methylates RNA 5' end to complete capping Myc Premature termination factors DCP2 Decaps nascent RNA for XRN2 digestion Dcp1a/Edc3 Microprocessor Cleaves hairpin structure for XRN2 digestion Tat, Senx XRN2 Torpedoes Pol II with RNA 5'-3' exonucleation TTF2 Releases Pol II from DNA Gdown1 GDOWN1 Antitermination and stabilizes paused Pol II TFIIF, Mediator Histone chaperone FACT H2A-H2B eviction and chaperone Tracks with Pol II NAP1 H2A-H2B chaperone RSC, CHD SPT6 H3-H4 chaperone Tracks with Pol II ASF1 H3-H4 chaperone H3K56ac Chromatin remodeler RSC SWI/SNF remodeling in gene body H3K14ac CHD1 Maintains gene body nucleosome organization FACT, DSIF NURF ISWI remodeling at promoter GAGA factor Poly(ADP-ribose) polymerase PARP Transcription independent nucleosome loss Tip60 Polymerase-associated factor complex PAF Loading dock for elongation factors SEC, FACT Histone tail modifiers MOF Acetylates H4K16 and recruits Brd4 H3S10ph, 14-3-3 TIP60 Acetylates H2AK5 and activates PARP Elongator Acetylates H3 and facilitates nucleosomal elongation Also in cytoplasm Rpd3C (Eaf3) Deacetylates and inhibits spurious initiation in gene body H3K36me3 SET1 Methylates H3K4 MLL/COMPASS SET2 Methylates H3K36 and regulates acetylation-deacetylation cycle Rpd3C PIM1 Phosphorylates H3S10 and recruits 14-3-3 and MOF RNF20/40 Monoubiquitinates H2BK123 and facilitates nucleosomal DNA unwrapping UbcH6, PAF - “Promoter-proximal pausing of RNA polymerase II (Pol II) precedes transcription elongation, but how Pol II is restrained is unknown. Chen et al. discovered that depletion of Pol II-associated factor 1 (PAF1) in human and fly cells resulted in redistribution of Pol II from promoter-proximal regions to gene bodies in thousands of genes; this was more pronounced in highly paused genes (indicating a strong dependence on PAF1 for pausing) and was accompanied by an increase in the proportion of elongating Pol II, which is phosphorylated on Ser2, at these genes. Ser2 phosphorylation and release from pausing upon PAF1 depletion were mediated by the recruitment to promoter-proximal regions of the super elongation complex (SEC), which includes the Pol II-activating kinase P-TEFb. This indicates that PAF1 restricts the access of SEC to promoters” (Zlotorynski 2015, doi:10.1038/nrm4053).
- “Paused RNA polymerase II (Pol II) that piles up near most human promoters is the target of mechanisms that control entry into productive elongation. Whether paused Pol II is a stable or dynamic target remains unresolved. We report that most 5′ paused Pol II throughout the genome is turned over within 2 min. ... We propose that Pol II occupancy near 5′ ends is governed by a cycle of ongoing assembly of preinitiated complexes that transition to pause sites followed by eviction from the DNA template. This model suggests that mechanisms regulating the transition to productive elongation at pause sites operate on a dynamic population of Pol II that is turning over at rates far higher than previously suspected. We suggest that a plausible alternative to elongation control via escape from a stable pause is by escape from premature termination” (Erickson, Sheridan, Cortazar and Bentley 2018, doi:10.1101/gad.316810.118)
-
Alternative coding sequences (transcription start and termination)
[This section covers alternative transcription start and end sites, and the occurrence of multiple coding sequences (open reading frames) in the same mRNA. These topics tend to bridge “decision-making during transcription” and “post-transcriptional decision-making”.]
Closely related to complexities of promoter architecture (see “Promoters” above): genes can have alternative
start sites, as well as alternative termination sites. This can result
in inclusion or elimination of protein-coding sections of a gene, and
therefore in different proteins. But it can also result in different
5'-UTR and 3'-UTR (untranslated regions) for a gene, with various
regulatory effects. These effects include life expectancy of the mRNA
and its localization within the cell for purposes of translation.
(Regarding 3'-UTR regions, see also
“Alternative cleavage,
polyadenylation, and deadenylation” below.)
“Far from acting as a constitutive mechanism to separate TUs
[transcription units, or genes] across the genome, termination can be
seen as an intricate process that displays remarkable flexibility and
regulatory potential. At the beginning of the gene, termination regulates
transcript release into productive elongation. [That is, termination
processes may prevent elongation.] It also acts as a checkpoint to
prevent the synthesis of defective mRNA, which could be translated into a
toxic (dominant negative) protein. At the end of the gene, termination
dictates which mRNA isoform is formed by APA [alternative
polyadenylation], thereby conferring selective expression properties on
the mRNA. Finally, termination can be overridden to adjust cells to
stress conditions or to adapt cells into a more pliable host for viral
replication. It is likely that future analysis of the termination process
has yet more surprises in store”
(Proudfoot 2016, doi:10.1126/science.aad9926).
“Altering the boundaries of mRNA molecules can affect how long they
stay intact, cause them to produce different proteins, or direct them
or their protein products to different locations, which can have a
profound biological impact” (European Molecular Biology Laboratory
2013).
“Many studies focusing on single genes have shown that the choice of a
specific TSS [transcription start site] has critical roles during
development and cell differentiation and aberrations in alternative
promoter and TSS use lead to various diseases including cancer,
neuropsychiatric disorders, and developmental disorders” (Klerk and
’t Hoen 2015, doi:10.1016/j.tig.2015.01.001).
“We investigated cell type-dependent differences in exon usage of over 18
000 protein-coding genes in 23 cell types from 798 samples of the
Genotype-Tissue Expression Project. We found that about half of the
expressed genes displayed tissue-dependent transcript isoforms.
Alternative transcription start and termination sites, rather than
alternative splicing, accounted for the majority of tissue-dependent exon
usage. We confirmed the widespread tissue-dependent use of alternative
transcription start sites in a second, independent dataset, Cap Analysis
of Gene Expression data from the FANTOM consortium. Moreover, our results
indicate that most tissue-dependent splicing involves untranslated exons
and therefore may not increase proteome complexity”
(Reyes and Huber 2018, doi:10.1093/nar/gkx1165).
Cryptic transcripts:
“Genomes are pervasively transcribed, producing a wide diversity
of coding and noncoding RNAs ... However, it remains unclear which
fraction of these transcripts exerts a biological role (direct or
regulatory). This question is particularly difficult to address when
these transcriptional units arise within, or in close proximity to,
protein coding genes in the same strand. Thus, their transcription
signals are difficult to distinguish from the nearby or even overlapped
protein coding genes. Among pervasively produced transcripts, so-called
cryptic transcripts constitute a particularly heterogeneous group.
Cryptic transcription is typically defined as the production of
noncanonical transcripts of unknown function”
(Wei, Hennig, Wang et al. 2019, doi:10.1101/gr.243378.118).
“We show that TSSs [transcription start sites] of chromatin-sensitive
internal cryptic transcripts retain comparable features of canonical TSSs
in terms of DNA sequence, directionality, and chromatin accessibility. We
define the 5′ and 3′ boundaries of cryptic transcripts and show that,
contrary to RNA degradation–sensitive ones, they often overlap with the
end of the gene, thereby using the canonical polyadenylation site, and
associate to polyribosomes. We show that chromatin-sensitive cryptic
transcripts can be recognized by ribosomes and may produce truncated
polypeptides from downstream, in-frame start codons ... Our work
suggests that a fraction of chromatin-sensitive internal cryptic
promoters initiates the transcription of alternative truncated mRNA
isoforms. The expression of these chromatin-sensitive isoforms is
conserved from yeast to human, expanding the functional consequences of
cryptic transcription and proteome complexity”
(Wei, Hennig, Wang et al. 2019, doi:10.1101/gr.243378.118).
“RNA polymerase II (Pol II) transcribes hundreds of thousands of
transcription units – a reaction always brought to a close by its
termination. Because Pol II transcribes multiple gene types, its
termination occurs in a variety of ways, with the polymerase being
responsive to different inputs. Moreover, it is not just a default
process occurring at the end of genes. Promoter-proximal and premature
termination is common and might in turn regulate gene expression levels.
Although some transcription termination mechanisms have been debated for
decades, research is only just underway on emergent processes”
(Eaton and West 2020, doi:10.1016/j.tig.2020.05.008).
“It has been recently shown that many proteins are lacking from reference
databases used in mass spectrometry analysis, due to their translation
templated on alternative open reading frames. This questions our current
understanding of gene annotation and drastically expands the theoretical
proteome complexity. The functions of these alternative proteins
(AltProts) still remain largely unknown ... [Our] study strongly
suggests novel roles of AltProts in multiple essential cellular functions
and supports the importance of considering them in future biological
studies” (Cardon, Franck, Coyaud et al. 2020, doi:10.1093/nar/gkaa277).
- The “fundamental concept of a single CDS [coding sequence, or open reading frame] is being challenged by increasing experimental evidence indicating that annotated proteins are not the only proteins translated from mRNAs. In particular, mass spectrometry (MS)-based proteomics and ribosome profiling have detected productive translation of alternative open reading frames. In several cases, the alternative and annotated proteins interact. Thus, the expression of two or more proteins translated from the same mRNA may offer a mechanism to ensure the co-expression of proteins which have functional interactions. Translational mechanisms already described in eukaryotic cells indicate that the cellular machinery is able to translate different CDSs from a single viral or cellular mRNA” (Mouilleron, Delcourt and Roucou 2016, doi:10.1093/nar/gkv1218)
- While alternative splicing in mammals can yield a large number of transcript variants from a given gene (see “Alternative Splicing” below), a study of cerebellar cells from mice found that alternative transcription produced even more variants than alternative splicing. This highlights “alternative promoters and transcriptional terminations as major sources of transcriptome diversity...Furthermore, the majority of genes associated with neurological diseases expressed multiple transcripts through alternative promoters, and we demonstrated aberrant use of alternative promoters in medullablastoma, cancer arising in the cerebellum” (Pal, Gupta, Kim et al. 2011).
- In yeast: “Hundreds of thousands of unique mRNA transcripts are generated from a genome of only about 8000 genes, even with the same genome sequence and environmental condition. ‘We knew that transcription could lead to a certain amount of diversity, but we were not expecting it to be so vast,’ explains Lars Steinmetz, who led the project. ‘Based on this diversity, we would expect that no yeast cell has the same set of messenger RNA molecules as its neighbour’”. This research on yeast has shown that “each gene could be transcribed into dozens or even hundreds of unique mRNA molecules, each with different boundaries”. “The researchers expect that such an extent of boundary variation will also be found in more complex organisms, including humans” (European Molecular Biology Laboratory 2013).
- Alternative transcription can affect either the protein-coding or the regulatory region of a gene. It can separate protein-coding regions previously thought to be conjoined, and can combine regions thought to have been independent (Pugh 2013; Pelechano, Wei and Steinmetz 2013). “The alternative promoter usage of TP73 results in two protein isoforms that perform opposing biological functions, and their balanced expression is a crucial factor in normal development and disease. In contrast, nine distinct mRNAs are produced from the BDNF gene through the use of alternative promoters, which differ in their 5'UTR [untranslated region at the 5' end of transcript] but translate the same protein. The distinct 5'UTRs function as the regulatory region responsible for the differential expression and localization of BDNF transcripts” (Pal, Gupta, Kim et al. 2011).
- “[We] identified 2035 mouse and 1847 human genes that utilize substantially distal novel 3' UTRs. Each of these extends at least 500 bases past the most distal 3' termini ... and collectively they add 6.6 Mb and 5.1 Mb to the mRNA space of mouse and human, respectively”. The alternatively cleaved and polyadenylated isoforms accumulated stably, and included transcripts “bearing exceptionally long 3' UTRs (many >10 kb and some >18 kb in length)”. Global tissue comparisons showed that the alternative cleaving and polyadenylation “were most prevalent in the mouse and human brain. Finally, these [3' UTR] extensions collectively contain thousands of conserved miRNA binding sites, and these are strongly enriched for many well-studied neural miRNAs. Altogether, these new 3' UTR annotations greatly expand the scope of post-transcriptional regulatory networks in mammals, and have particular impact on the central nervous system” (Miura, Shenker, Andreu-Agullo et al. 2013).
- “When RNA polymerase II (Pol II) reaches the gene end, it first slows down over the terminator. This is partly because 3'-end cleavage and polyadenylation (CPA complex is recruited onto Pol II when poly(A) signals appear in the nascent transcript. This nascent transcript will often invade the DNA duplex to form an R-loop structure, which induces further polymerase slowdown. During this time, CPA releases mRNA from chromatin into eventual cytoplasmic translation. Pol II continues to transcribe its DNA template after mRNA release. However, this is short-lived, as an exonuclease (Xrn2) degrades the transcript from its 5' end. When this molecular torpedo catches up with Pol II, then conformational shockwaves are transmitted into its active site, which releases Pol II from the DNA template. Pol II is then free to restart transcription on another gene promoter” (Proudfoot 2016, doi:10.1126/science.aad9926).
- “Recent studies reveal that cellular stress such as osmotic or heat shock, as well as viral infection or cancer-inducing mutations, can all promote aberrant termination. Under these varied conditions, many genes fail to terminate transcription. The resulting extensive readthrough transcription can cause massive deregulation of downstream gene expression” (Proudfoot 2016, doi:10.1126/science.aad9926).
- “Dhir et al. now find that transcription termination of lncRNA [long noncoding RNA] transcripts containing primary miRNAs (lnc-pri-miRNAs) — which encode 17.5% of human miRNAs — involves cleavage by the Microprocessor complex rather than the canonical cleavage and polyadenylation pathway. The Microprocessor complex, which comprises the double-stranded RNA-binding protein DGCR8 and the RNase III endonuclease Drosha, is known to process pri-miRNA-containing protein-coding transcripts to give rise to miRNAs. Here, the authors found that liver-specific lnc-pri-miR-122 is not polyadenylated but contains a cleavage site for Drosha at its 3ʹ end. Moreover, depletion of DGCR8 or Drosha led to readthrough transcription, indicative of a termination defect. Genome-wide chromatin RNA sequencing analyses in HeLa cells indicated that Microprocessor terminates transcription of most lnc-pri-miRNAs” (Baumann 2015, doi:10.1038/nrm3976 — reporting on work by Dhir et al. 2015, doi:10.1038/nsmb.2982).
- A study of yeast genes showed that differences in the 5'-UTR [the untranslated region proximal to the transcription start site] often affected rates of protein production (translation), with rates varying up to 100-fold. “Because transcription start site heterogeneity is common, we suggest that transcription start site choice is greatly under-appreciated as a quantitatively significant mechanism for regulating protein production” (Rojas-Duran and Gilbert 2012).
- In higher organisms, transcripts resulting from alternative transcription are often also alternatively spliced.
- “As Pol II [RNA polymerase II] is coupled with RNA 3'-end processing, the timing of Pol II release can also dictate the length of the final RNA product and thus affect the stability, localization and ultimate functionality of nascent transcripts” (Kuehner, Pearson and Moore 2011).
- Circadian clock-related transcription (and other?) factors interact with alternative transcription start sites, “leading to rhythmic expression of some isoforms but not others” (Edery 2011).
- There are “at least two major classes of promoters in vertebrates, and these differ substantially in the signals they use for TSS [transcription start site] selection”. The two classes have either high or low CG content. The high CG content of the one class tends to be associated with a nucleosome-free region and broadly distributed TSSs, and it “can in itself prime the promoter for transcription activation through chromatin signals”. By contrast, the low-CG promoters “seem to follow a regulatory logic more akin to the classic view of transcription initiation where the promoter is inactive by default, but can be activated by specific TFs [transcription factors] which recruit chromatin remodeling complexes, which in turn remove the nucleosome to expose more TFBSs [transcription factor binding sites] or the TSS”. Any given promoter may exhibit a blend of these (and other related) features (Valen and Sandelin 2011).
- “Alternative polyadenylation (APA) generates mRNA isoforms with 3' untranslated regions (UTRs) of different lengths; longer 3' UTRs contain regulatory elements that affect mRNA localization and mRNA and protein abundance. Berkovits and Mayr now show that APA can also regulate protein localization, independent of mRNA localization” (Zlotorynski 2015, doi:10.1038/nrm3996).
- “To better understand the gene regulatory mechanisms that program developmental processes, we carried out simultaneous genome-wide measurements of mRNA, translation, and protein through meiotic differentiation in budding yeast. Surprisingly, we observed that the levels of several hundred mRNAs are anti-correlated with their corresponding protein products. We show that rather than arising from canonical forms of gene regulatory control, the regulation of at least 380 such cases, or over 8% of all measured genes, involves temporally regulated switching between production of a canonical, translatable transcript and a 5′ extended isoform that is not efficiently translated into protein. By this pervasive mechanism for the modulation of protein levels through a natural developmental program, a single transcription factor can coordinately activate and repress protein synthesis for distinct sets of genes. The distinction is not based on whether or not an mRNA is induced but rather on the type of transcript produced” (Cheng, Otto, Powers et al. 2018, doi:10.1016/j.cell.2018.01.035).
-
Overlapping and interleaved transcription
“In gene-rich regions, both strands of DNA are often pervasively
transcribed. Transcription occurs upstream, downstream, and antisense to
genes and may span several genes. Pervasive transcription has the
potential to activate or repress neighbouring genes by altering DNA
supercoiling or changing the structure and composition of the chromatin
... An interleaved genome is highly plastic. Altering gene expression at
one gene in cluster can result in a new functional transcription unit
over a different region of the cluster”
(Mellor, Woloszczuk and Howe 2016, doi:10.1016/j.tig.2015.10.006).
“Eukaryotic genomes are pervasively transcribed but until recently this
noncoding transcription was considered to be simply noise. Noncoding
transcription units overlap with genes and genes overlap other genes,
meaning genomes are extensively interleaved. Experimental interventions
reveal high degrees of interdependency between these transcription units,
which have been co-opted as gene regulatory mechanisms. The precise
outcome depends on the relative orientation of the transcription units
and whether two overlapping transcription events are contemporaneous or
not, but generally involves chromatin-based changes. Thus transcription
itself regulates transcription initiation or repression at many regions
of the genome”
(Mellor, Woloszczuk and Howe 2016, doi:10.1016/j.tig.2015.10.006).
-
Post-translational modifications of RNA Polymerase
“The C-terminal domain (CTD) of RNA polymerase II (Pol II) consists of
conserved heptapeptide repeats that function as a binding platform for
different protein complexes involved in transcription, RNA processing,
export, and chromatin remodeling. The CTD repeats are subject to
sequential waves of posttranslational modifications during specific
stages of the transcription cycle. These patterned modifications have
led to the postulation of the ‘CTD code’ hypothesis, where
stage-specific patterns define a spatiotemporal code that is recognized
by the appropriate interacting partners” (Zhang, Rodríguez-Molina,
Tietjen et al. 2012).
- During transcription, RNA polymerase II undergoes changing combinations of post-translational modifications of its carboxy-terminal domain [CTD]. These play a role in the transition from stage to stage of transcription, from initiation to pausing to elongation to termination. As RNA polymerase II is recruited to gene promoters, its CTD is largely unphosphorylated. But as transcription proceeds, some serine residues are progressively phosphorylated and others dephosphorylated, and thereby “drive the transcription cycle and regulate cotranscriptional events”. For example, “Ser5 phosphorylation by the general transcription factor TFIIH results in the dissociation of the initiation-specific Mediator complex from RNAPII, helping to release the polymerase from the promoter. Ser5 phosphorylation also helps recruit specific factors to the transcribed gene, including the mRNA 5'-end capping enzyme, chromatin-modifying factors, and mRNA splicing factors. As the transcription cycle proceeds into elongation, Ser5 phosphorylation is removed by CTD phosphatases, and another mark, Ser2 phosphorylation, is added in its place. Among its known functions, Ser2 phosphorylation plays an important role in attracting histone-modifying enzymes, as well as in mRNA 3'-end processing”. Threonine residues also go through cycles of phosphorylation and dephosphorylation, playing “a general and important role in transcript elongation in mammalian cells” (Svejstrup 2012).
- Combining this with the note above about pausing during elongation and its implications for chromatin remodeling: It appears that RNA polymerase II both rides a wave of chemical modifications as it transits a gene, and in turn plays a role in modulating that wave. It helps to modify histones ahead of its movement, and to restore histone states behind itself, even as its own behavior is affected by those histones and their modifications.
- Among the other modifications of the carboxy-terminal domain of RNA polymerase II in mammals: it is hypothesized that methylation of an arginine residue may play a role in targeting RNA polymerase II to distinct types of genes. In particular, it appears to help regulate expression of snRNA (small nuclear RNA) and snoRNA (small nucleolar RNA) (Sims, Rojas, Beck et al. 2011).
- Of course, the enzymes applying these post-translational modifications must somehow themselves perform in a well-regulated manner.
- “In addition to its many roles in transcription initiation, elongation, and termination, the CTD has been implicated in a variety of transcription-extrinsic processes, such as mRNA export and stress response” (Zhang, Rodríguez-Molina, Tietjen et al. 2012).
-
Chromosome looping
Various evidences suggest that “polymerases might be the molecular
ties maintaining [chromosome] loops” (Papantonis and Cook 2010). (See
also “Chromosome looping and
long-distance chromatin interaction” under
“THREE-DIMENSIONAL ORGANIZATION OF CHROMOSOMES,
NUCLEUS, AND CELL”, below.)
-
RNA polymerase and alternative splicing
See “Role of RNA polymerase” under “Alternative Splicing below. -
Transcription and formation of G-quadruplexes
Transcription at one DNA locus can cause the formation of G-quadruplex structures thousands of base pairs upstream from the transcription, with implications for gene expression. See “DNA G-quadruplexes” under “OTHER ASPECTS OF THE MOLECULAR STRUCTURE OF DNA AND RNA”, below.) - See also “mRNA coordinators” under POST-TRANSCRIPTIONAL DECISION-MAKING.
-
5'-end cap, and cap-binding proteins
mRNAs and some other mRNAs receive a “cap” at their “front” (5') end early
in the transcription process. This consists of a methylated guanine
nucleotide with an unusual linkage. The next nucleotide or two (at the
beginning of the original transcription product) may also be methylated,
thereby contributing to the cap structure. The cap helps to prevent
unwarranted degradation of the mRNA by enzymes that could otherwise “eat
away” at the 5' end. It also facilitates proper attachment of the mRNA to
ribosomes. Other functions, some of which are documented here, are
mediated by protein complexes that bind to the cap.
There are several distinct nuclear cap-binding complexes (CBCs), which have
some core subunits in common.
Cap-binding complexes are “at the center of an RNA-surveillance process
that can couple multiple steps of transcription and RNA processing and
thereby determine the fate of nascent Pol II transcripts. ... The extent of
coupling among discrete events has been underappreciated” (Müller-McNicoll
and Neugebauer 2014).
The following abstract merely hints at some of the many aspects of RNA
cap-binding, almost none of which are discussed in the brief notes below:
“The largely nuclear cap-binding complex (CBC) binds to the 5′ caps of RNA
polymerase II (RNAPII)-synthesized transcripts and serves as a dynamic
interaction platform for a myriad of RNA processing factors that regulate
gene expression. While influence of the CBC can extend into the cytoplasm,
here we review the roles of the CBC in the nucleus, with a focus on
protein-coding genes. We discuss differences between CBC function in yeast
and mammals, covering the steps of transcription initiation, release of
RNAPII from pausing, transcription elongation, cotranscriptional pre-mRNA
splicing, transcription termination, and consequences of spurious
transcription. We describe parameters known to control the binding of
generic or gene-specific cofactors that regulate CBC activities depending on
the process(es) targeted, illustrating how the CBC is an ever-changing
choreographer of gene expression”
(Rambout and Maquat 2020, doi:10.1101/gad.339986.120).
- One of the cap-binding complexes is known as “CBCA”: “For short transcripts, such as snRNAs [small nuclear RNAs] and histone mRNAs, CBCA promotes degradation when Pol II reads [past the site]. For long mRNAs, the opposite is true: CBCA promotes decay when the mRNA is cleaved close to the cap and promotes export when the transcript is correctly processed at the 3' end” (Müller-McNicoll and Neugebauer 2014). How all the proper “decisions” are made, based on transcript length, the nature of the RNA, and interaction with other vital processes (see next bullet item), is rather hard to fathom. Presumably a very complicated story will progressively unfold.
- The “sorting and surveillance activities mediated by CBCA take place during transcription and compete with other RNA-processing events, such as RNA editing and splicing, in a cotranscriptional race against time” (Müller-McNicoll and Neugebauer 2014).
- Another CBC complex is “CBCN”, which acts on three RNA classes: “misprocessed 3'-extended snRNAs, histone mRNAs and PROMPTs. PROMPTs are noncoding transcripts that are capped and polyadenylated and are generated through transcription at most bidirectional promoters and 3' processing at promoter-proximal polyadenylation sites”. CBCN seems to play a role in degradation of antisense PROMPTs and misprocessed transcripts (Müller-McNicoll and Neugebauer 2014). (It is always good to bracket in your mind such terms as ‘misprocessed’, which often mean, in effect, “Processed according to functions not yet discovered”.)
-
“Competition of the hMTR4 helicase and the mRNA export adaptor ALYREF for
interaction with the nuclear cap‐binding complex determines the
specificity of exosome recruitment to nuclear RNAs, forming a checkpoint
to ensure that only functional mRNAs and lncRNAs are exported to the
cytoplasm”. More specifically:
- “Disruption of mRNA processing and export in human cells triggers significant mRNA and lncRNA degradation by the nuclear exosome”.
- “hMTR4 triggers RNA decay by directing the nuclear exosome to specific targets”.
- “hMTR4 and export receptor ALYREF compete for the binding to the nuclear cap‐binding complex”.
- “The competition between hMTR4 and ALYREF determines if nuclear RNA pools are destined for degradation or export”.
-
Histone modifications
Some histone modifications, unlike those discussed under PRE-TRANSCRIPTIONAL
DECISION-MAKING above, occur
during transcription. “Dynamic incorporation of histones into nucleosomes
over the body of genes regulates the process of gene transcription”
(Venkatesh, Smolle, Li et al. 2012).
- As an example: methylation of H3K36 occurs co-transcriptionally. This modification (1) targets and activates a deacetylase complex, and (2) suppresses the interaction between histone H3 and histone chaperones that otherwise would enhance histone exchange and the incorporation of acetylated histones into the gene body. By down-regulating acetylation in these ways, the methylation of H3K36 helps to suprress spurious cryptic transcripts originating from sites within the gene coding region. This illustrates the kind of meaningful interplay that can occur among the various histone tail modifications (Venkatesh, Smolle, Li et al. 2012).
- “Long non-coding RNA (lncRNA) transcription into a downstream promoter frequently results in transcriptional interference. However, the mechanism of this repression is not fully understood. We recently showed that drug tolerance in fission yeast Schizosaccharomyces pombe is controlled by lncRNA transcription upstream of the tgp1+ permease gene. Here we demonstrate that transcriptional interference of tgp1+ involves several transcription-coupled chromatin changes mediated by conserved elongation factors Set2, Clr6CII, Spt6 and FACT. These factors are known to travel with RNAPII and establish repressive chromatin in order to limit aberrant transcription initiation from cryptic promoters present in gene bodies. We therefore conclude that conserved RNAPII-associated mechanisms exist to both suppress intragenic cryptic promoters during genic transcription and to repress gene promoters by transcriptional interference ... Given that eukaryotic genomes are pervasively transcribed, transcriptional interference likely represents a more general feature of gene regulation than is currently appreciated” (Ard and Allshire 2016, doi:10.1093/nar/gkw801).
-
Transcription of noncoding RNAs
- Transcription of noncoding RNAs in intergenic regions of the genome generates “transcription ripples” that propagate considerable distances downstream, activating protein-coding genes up to 100 kilobases away (Ebisuya, Yamamoto, Nakajima and Nishida 2008; Carninci 2008).
- There are also suggestions that transcription of noncoding RNA can cause direct “transcriptional interference,” negatively regulating neighboring genes (Dinger, Amaral, Mercer and Mattick 2009).
- “RNA polymerase II (RNAP II) non-coding transcription is now known to cover almost the entire eukaryotic genome, a phenomenon referred to as pervasive transcription. As a consequence, regions previously thought to be non-transcribed are subject to the passage of RNAP II and its associated proteins for histone modification. This is the case for the nucleosome-depleted regions (NDRs), which provide key sites of entry into the chromatin for proteins required for the initiation of coding gene transcription and DNA replication. In this review, recent data on the effects of pervasive transcription through NDRs are summarized and a model is proposed to explain how RNAP II-driven transcription is able to modify the nucleosomes flanking the NDRs, leading to nucleosome repositioning and NDR closure. Even though much of the mechanistic detail underlying these events remains to be elucidated, such a model provides a basis to explain how non-coding transcription through NDRs can regulate the initiation of coding gene expression and DNA replication” (Liu and Cai 2019, doi:0.1002/bies.201900159).
-
Riboswitches and regulatory 5' untranslated regions (5'UTRs)
- “Our findings suggest that the number and diversity of pathways regulated by r5'UTRs [regulatory 5' untranslated regions] has been underestimated” (Livny and Waldor 2010).
-
RNA folding
It is not only proteins whose complex folding affects their
functioning. RNA transcripts also need to fold appropriately in
order to be spliced, edited, and translated properly. This folding of
an RNA transcript can be affected by the speed of its transcription.
And so factors affecting this speed can determine the outcome of gene
expression. (See also RNA structure,
below.)
“Evidence gathered in recent years is consolidating our understanding that
posttranscriptional regulation contributes as much and probably more than the
better-characterized transcriptional regulation to determine gene expression”
(Dominissini 2014).
“A network of proteins regulates the expression of messenger RNA (mRNA) to
maintain homeostasis and adapt cell physiology to changing environments1. This
network includes cis-acting mRNA sequence elements and
trans-acting factors that bind the transcript to regulate its fate2.
RNA-binding proteins (RBPs) determine whether an mRNA is translationally
activated or repressed, localized to a specific region within the cell, or
degraded1. RBPs can also remodel RNA structure and act as chaperones to prevent
RNA aggregation3,4. Determining the effects of regulatory RBPs is critical to
understanding post-transcriptional control of gene expression”
(Reynaud, McGeachy, Noble et al. 2023; doi:10.1038/s41594-023-00999-5).
-
Creation of mRNA variants
“Most human genes encode a repertoire of mRNA variants generated by
alternative splicing, alternative polyA [polyadenylation] site selection,
editing, and selection of alternative first exons. Eighty percent of
alternative splicing affects the open reading frame to produce diverse
protein isoforms or introduce premature termination codons to affect mRNA
levels by nonsense-mediated decay. Variability within untranslated regions
affects cis-acting elements that regulate translation efficiency,
mRNA stability or mRNA localization. A large fraction of mRNA complexity
is regulated in response to changing physiological needs, which results in
a highly versatile and dynamic proteome” (Cooper 2011).
-
RNA splicing
- Splicing is a way of modifying the RNA molecules produced by transcription. Certain sections of the RNA (introns) are removed, and the remaining sections (exons) are stitched together. The underlying DNA sequence provides certain short sequences that, in the transcribed RNA, act as signals for the spliceosomes (RNA-protein complexes) that, in many cases, perform the splicing.
- The spliceosome is “composed of five small nuclear ribonucleoproteins (snRNPs) and more than 50 non-snRNPs, which recognize and assemble on exon-intron boundaries to catalyse intron processing of the pre-mRNA” (Keren, Lev-Maor and Ast 2010).
- More rarely, self-splicing introns can excise themselves from the RNA transcript, using one of at least two different methods. The introns are classified as Group I or Group II introns, depending on the method. A third group is rather more hazily defined.
- Yet another, altogether different splicing process is applied to tRNA molecules.
- “The list of developmentally and tissue-restricted splicing factors is larger than previously thought,” but it’s not yet known how extensive this specificity is (Tavanez and Valcárcel 2010).
- Regarding kinetic aspects of splicing: “First, spliceosome assembly is highly ordered, indicating that, although factors such as the U1-snRNPs and U2-snRNPs bind and unbind on the time scale of seconds to minutes, there is a directionality to the process that is reinforced by the consumption of ATP. Second, commitment to splicing does not occur through a single irreversible step but rather is the cumulative outcome of many coupled reactions. As a consequence, no single kinetic step dominates the reaction, and the net rate of splicing is due to many sequential kinetic steps. For these in vitro studies, the time from U1-snRNP binding to intron removal was measured to be ∼12 min. One of the primary conclusions of this single-molecule analysis is that spliceosome assembly and pre-mRNA splicing are reversible at almost every step, which opens up the possibility of regulation at multiple points. Subsequent work using this same approach indicates that the order of assembly of the spliceosome can follow slightly different routes and still result in the same pre-mRNA splicing outcome. Thus, there is a considerable plasticity to the spliceosome” (Chen and Larson 2016, doi:10.1101/gad.281725.116).
- Nuclear speckles are “defined by high concentrations of protein and noncoding RNA regulators of pre-mRNA splicing3 ... Here we show that genes localized near nuclear speckles display higher spliceosome concentrations, increased spliceosome binding to their pre-mRNAs and higher co-transcriptional splicing levels than genes that are located farther from nuclear speckles. Gene organization around nuclear speckles is dynamic between cell types, and changes in speckle proximity lead to differences in splicing efficiency. Finally, directed recruitment of a pre-mRNA to nuclear speckles is sufficient to increase mRNA splicing levels. Together, our results integrate the long-standing observations of nuclear speckles with the biochemistry of mRNA splicing and demonstrate a crucial role for dynamic three-dimensional spatial organization of genomic DNA in driving spliceosome concentrations and controlling the efficiency of mRNA splicing” (Bhat, Chow, Emert et al. 2024, doi:10.1038/s41586-024-07429-6).
-
Alternative splicing
Splicing of the same pre-mRNA molecule can often be done in different
ways, yielding different proteins. This alternative splicing is, in
part, regulated by specific sequences on the pre-mRNA molecules
together with a ribonucleoprotein complex (the spliceosome) that acts
on the pre-mRNA. Different combinations of proteins and their
interactions with the RNA sequences lead to different splicing results.
But in recent years a number of additional factors have been (and are
still being) found to bear on splicing.
“Fundamental differences in splicing patterns have been observed in
epithelial versus mesenchymal cells, neurons before and after
depolarization, heart tissue during development and in disease
conditions, resting and activated T cells, cells during circadian
rhythms and cells before and after initiation of apoptosis, to name but
a few examples” (Heyd and Lynch 2011). “Protein isoforms produced by
alternative promoters or alternative splicing can have subtle or even
opposing functional differences that can, in turn, have profound
biological consequences” (Weatheritt and Gibson 2012). “Even
relatively modest changes in alternative splicing can have dramatic
consequences, including altered cellular responses, cell death, and
uncontrolled proliferation that can lead to disease” (Luco and Misteli
2011).
“RNA-binding proteins (RBPs) regulate alternative splicing through their
expression level, intracellular localization, activity and, in some
cases, their own alternative splicing. RBPs promote or inhibit the
recognition of alternative regions by the spliceosome machinery. Multiple
RBPs can regulate alternative splicing in a cooperative or competitive
manner and also exert their regulatory functions by coupling with other
splicing regulators (such as the transcriptional machinery or epigenetic
readers). The expression levels of RBPs are tightly regulated during
organ development and cell differentiation. Tissue- and
cell-type-specific RBP expression patterns give rise to different
splicing products. Therefore, the modulation of splicing transitions
through RBPs is one of the main mechanisms of splicing coordination,
particularly during development”
(Baralle and Giudice 2017, doi:10.1038/nrm.2017.27).
“Alternative splicing outcomes can be modulated also by other means,
including transcription and epigenetic changes. It has been shown that
RNA polymerase II (Pol II) elongation rates, which are controlled by
chromatin modifications, DNA methylation and nucleosome occupancy, can
greatly influence splicing. When elongation rates are reduced, weak
splice sites are better recognized and alternative exons tend to be
included; by contrast, when Pol II elongation is enhanced, the
recognition of weak splice sites is impaired and alternative exons tend
to be skipped. However, slow Pol II can favour the recruitment of
specific RBPs that promote exon inclusion (positive regulators) or
skipping (negative regulators), whereas fast Pol II hampers that
recruitment. Increased nucleosome occupancy, Pol II pausing, DNA
methylation and specific histone marks at exons relative to introns
support the idea that these epigenetic signatures can also help the
splicing machinery to recognize alternative exons. Indeed, global
correlations are being demonstrated between splicing patterns and
specific histone marks, DNA-methylation patterns, nucleosome occupancy,
Pol II positioning and RBP binding”
(Baralle and Giudice 2017, doi:10.1038/nrm.2017.27).
All this has remarkably transformed the old idea that genes
determine proteins in any straightforward way. It’s not a matter of
gene “control” at all: “The emerging evidence places alternative
splicing in a central position in the flow of eukaryotic genetic
information, between transcription and translation, in that it can
respond not only to various signalling pathways that target the
splicing machinery but also to transcription factors and chromatin
structure” (Kornblihtt, Schor, Alló et al. 2013).
“Although branchpoint recognition is an essential component of intron
excision during the RNA splicing process, the branchpoint itself is
frequently assumed to be a basal, rather than regulatory, sequence
feature. However, this assumption has not been systematically tested due
to the technical difficulty of identifying branchpoints and quantifying
their usage. Here, we analyzed ∼1.31 trillion reads from 17,164 RNA
sequencing data sets to demonstrate that almost all human introns contain
multiple branchpoints. This complexity holds even for constitutive
introns, 95% of which contain multiple branchpoints, with an estimated
five to six branchpoints per intron. Introns upstream of the highly
regulated ultraconserved poison exons of SR genes contain twice as many
branchpoints as the genomic average. Approximately three-quarters of
constitutive introns exhibit tissue-specific branchpoint usage. In an
extreme example, we observed a complete switch in branchpoint usage in
the well-studied first intron of HBB (β-globin) in normal bone marrow
versus metastatic prostate cancer samples. Our results indicate that the
recognition of most introns is unexpectedly complex and tissue-specific
and suggest that alternative splicing catalysis typifies the majority of
introns even in the absence of differences in the mature mRNA”
(Pineda and Bradley 2018, doi:10.1101/gad.312058.118).
“In nervous systems, alternative splicing has emerged as a fundamental
mechanism not only for the diversification of protein isoforms but also
for the spatiotemporal control of transcripts. Thus, alternative splicing
programs play instructive roles in the development of neuronal cell
type–specific properties, neuronal growth, self-recognition, synapse
specification, and neuronal network function”
(Furlanis and Scheiffele 2018, doi:10.1146/annurev-cellbio-100617-062826)
“Four UBLs, namely, ubiquitin, SUMO, Hub1, and Sde2, are involved in
eukaryotic pre-mRNA splicing. They modify the spliceosomes and promote
splicing by adding new surfaces for intermolecular interactions, thereby
refining the outcome of gene expression. In this review article, we
highlight recent discoveries with an emphasis on the emerging roles of
UBLs in splicing regulation”
(Chanarat and Mishra 2018, doi:10.1016/j.tibs.2018.09.001).
“During the splicing reaction, the dynamic spliceosome has an immobile
core of about 20 protein and RNA components, which are organized around a
conserved splicing active site. The divalent metal ions, coordinated by
U6 small nuclear RNA (snRNA), catalyze the branching reaction and exon
ligation. The spliceosome also contains a mobile but compositionally
stable group of about 13 proteins and a portion of U2 snRNA, which
facilitate substrate delivery into the splicing active site. The
spliceosomal transitions are driven by the RNA-dependent
ATPase/helicases, resulting in the recruitment and dissociation of
specific splicing factors that enable the reaction. In summary, the
spliceosome is a protein-directed metalloribozyme.”
(Yan, Wan and Shi 2018, doi:10.1101/cshperspect.a032409 )
“Histone modifications and RNA splicing, two seemingly unrelated gene
regulatory processes, greatly increase proteome diversity and profoundly
influence normal as well as pathological eukaryotic cellular functions.”
Recent studies “reveal that HDACs [histone deacetylases] interact with
spliceosomal and ribonucleoprotein complexes, actively control the
acetylation states of splicing-associated histone marks and splicing
factors, and thereby unexpectedly could modulate splicing ... the
convergence of two parallel fields ... supports the argument that HDACs,
and perhaps most histone modifying enzymes, are much more versatile and
far more complicated than their initially proposed functions.
Analogously, an HDAC-RNA splicing connection suggests that splicing is
regulated by additional upstream factors and pathways yet to be defined
or not fully characterized. Some human diseases share common underlying
causes of aberrant HDACs and dysregulated RNA splicing and, thus, further
support the potential link between HDACs and RNA splicing”
(Rahhal and Seto 2019, 10.1093/nar/gkz292).
“Changes in the rate of transcription can influence RNA splicing, but
whether splicing influences transcription start site (TSS) location and
activity was not known. Fiszbein et al. describe a novel process called
exon-mediated activation of transcription starts (EMATS), in which
splicing of internal exons can enhance gene expression through the
activation of proximal TSSs ... the group found that inhibiting the
splicing of [certain exons] reduced levels of nascent RNA for each gene.
This finding suggested that splicing of the exon had an impact on
upstream transcription initiation. Genes with [evolutionarily] new exons
tended to have multiple TSSs and were enriched for TSSs located just
upstream of the exon”. Indications are that a new exon “mediates
recruitment of transcriptional machinery to the proximal TSS ... This
effect was not exclusive to new exons”
(Willson 2020, doi:10.1038/s41576-019-0207-2).
In an article titled, “Distinct p53 Isoforms Code for Opposing
Transcriptional Outcomes”:
“we investigate synthetic and native cis-regulatory elements in
Drosophila to examine opposing features of p53-mediated
transcriptional control in vivo. We show that transcriptional
repression by p53 operates continuously through canonical DNA binding
sites that confer p53-dependent transactivation at earlier developmental
stages. p53 transrepression is correlated with local H3K9me3 chromatin
marks and occurs without the need for stress or Chk2. In sufficiency
tests, two p53 isoforms qualify as transrepressors and a third qualifies
as a transcriptional activator. Targeted isoform-specific knockouts
dissociate these opposing transcriptional activities, highlighting
features that are dispensable for transactivation but critical for
repression and for proper germ cell formation. Together, these results
demonstrate that certain p53 isoforms function as constitutive
tissue-specific repressors, raising important implications for tumor
suppression by the human counterpart”
(Wylie, Jones, Das et al. 2022, doi:10.1016/j.devcel.2022.06.015).
“Glucose is the main source of energy for cells. In this issue of Cell, a
study now shows that glucose has additional non-energetic functions,
acting as a biomolecular cue that regulates alternative splicing during
epidermal differentiation. As keratinocytes differentiate, glucose
associates with RNA-binding protein DDX21 and modulates its interaction
properties, which modifies splicing decisions”
(Carmo-Fonseca 2023, doi:10.1016/j.cell.2022.11.025).
“Alternative splicing is a substantial contributor to the high complexity
of transcriptomes of multicellular eukaryotes. In this Review, we discuss
the accumulated evidence that most of this complexity is reflected at the
protein level and fundamentally shapes the physiology and pathology of
organisms. This notion is supported not only by genome-wide analyses but,
mainly, by detailed studies showing that global and gene-specific
modulations of alternative splicing regulate highly diverse processes
such as tissue-specific and species-specific cell differentiation,
thermal regulation, neuron self-avoidance, infrared sensing, the Warburg
effect, maintenance of telomere length, cancer and autism spectrum
disorders (ASD)”
(Marasco and Kornblihtt 2023, doi:10.1038/s41580-022-00545-z).
“Alternative splicing affects more than 95% of multi-exon genes in the
human genome. These changes affect the proteome in a myriad of ways.
Here, we review our understanding of the breadth of these changes from
their effect on protein structure to their influence on interactions.
These changes encompass effects on nucleic acid binding in the nucleus to
protein–carbohydrate interactions in the extracellular milieu, altering
interactions involving all major classes of biological molecules. Protein
isoforms have profound influences on cellular and tissue physiology, for
example, by shaping neuronal connections, enhancing insulin secretion by
pancreatic beta cells and allowing for alternative viral defense
strategies in stem cells. More broadly, alternative splicing enables
repurposing proteins from one context to another and thereby contributes
to both the evolution of new traits as well as the creation of
disease-specific interactomes that drive pathological phenotypes”.
“Alternative splicing [is] a central regulator of protein function with
implications for almost every biological process”
(Kjer-Hansen and Weatheritt 2023, doi:10.1038/s41594-023-01155-9).
- Recent studies indicate that 95–100% of human pre-mRNAs (precursor mRNAs) containing more than one exon are processed to yield multiple distinct mRNAs, called isoforms. “And not only do most genes encode pre-mRNAs that are alternatively spliced, but also the number of mRNA isoforms encoded by a single gene can vary from two to several thousand”. One gene in the fruit fly “can generate 38,016 distinct mRNA isoforms, a number far in excess of the total number of genes (~14,500) in the organism” (Nilsen and Graveley 2010).
- Further, “isoform expression by a gene does not follow a minimalistic expression strategy”. The tendency, rather, is “for genes to express many isoforms simultaneously, with a plateau at about 10–12 expressed isoforms per gene per cell line” (Djebali, Davis, Merkel et al. 2012).
- “As cells differentiate and respond to stimuli in the human body, over one million different proteins are likely to be produced from less than 25,000 genes” (de Almeida and Carmo-Fonseca 2012).
- Protein isoforms from a single gene can have differing functions. In fact, “a frequent outcome of alternative splicing is the production of proteins with opposing functions, a phenomenon illustrated perhaps most dramatically by the fact that a large majority of genes encoding proteins that function in apoptotic cell death pathways give rise to either pro- or anti-apoptotic isoforms by alternative splicing” (David and Manley 2010).
- The existence of opposing functionalities does not depend on production of two different proteins by alternative splicing. Two of the functions of an mRNA called FSTL1 depend instead on whether the transcript is spliced to produce a microRNA (miR-198) or a protein. Under normal conditions, miR-198 is produced, and it plays a key role in preventing cell migration. But when a wound occurs, the cell downregulates production of miR-198 and increases the production of an FSTL1 protein by splicing the transcript differently. This protein helps promote cell migration and therefore re-epithelialization and wound healing. In non-healing chronic diabetic ulcers, this changeover from the microRNA to the protein doesn’t happen, which can prevent healing and lead to the necessity for limb amputation (Sundaram, Common, Gopal et al. 2013).
- One study, focusing on T-cells of the human immune system, identified 178 exons in 168 genes that exhibited “robust changes in [exon] inclusion in response to stimulation” of the cells. There was global coordination of alternative splicing following this stimulation. “These signal-responsive exons are significantly enriched in genes with functional annotations specifically related to immune response. The vast majority of these genes also exhibit differential alternative splicing between naive and activated primary T cells. Comparison of the responsiveness of splicing to various stimuli in the cultured and primary T cells further reveals at least three distinct networks of signal-induced alternative splicing events. Importantly, we find that each regulatory network is specifically associated with distinct sequence features, suggesting that they are controlled by independent regulatory mechanisms” (Martinez, Pan, Cole et al. 2012).
- Alternative exons are enriched for the production of intrinsically disordered regions in proteins, and these in turn are enriched in short linear motifs (SLiMs, 3–10 amino acids in length) that can act as sites for post-translational modifications or as binding sites that target signaling and other molecules. There is “evidence that the removal, addition, or creation of SLiMs as small as 3 amino acids in length, by inclusion or exclusion of exons, leads to novel cellular localisation, resistance to cleavage, longer half-lives, novel partner binding, and altered binding affinities. This can result in novel and even opposing functions. On a pathway level, this can change the sensitivity of a pathway, disrupt branching of a pathway, weaken the stability of complexes, facilitate cooperativity, and even create opposing pathways” (Weatheritt and Gibson 2012).
- “Most typically, alternative splicing involves the differential inclusion or exclusion of a specific exon in different cell types or growth conditions, although all other imaginable patterns have been observed, including retention of introns, exclusion of a portion of an exon, and mutually exclusive inclusion of exons... Importantly, any of these differential patterns have the capacity to alter the open reading frame of the resultant mRNA or alter the presence of cis-regulatory elements that control mRNA stability or translation. Therefore, the precise control of alternative splicing plays an essential role in shaping the proteome of any given cell, and changes in splicing patterns can significantly alter cellular function in response to changing environmental conditions” (Heyd and Lynch 2011).
- “Intron retention is overwhelmingly perceived as an aberrant splicing event with little or no functional consequence. However, recent work has now shown that intron retention is used to regulate a specific differentiation event within the haematopoietic system by coupling it to nonsense-mediated mRNA decay. Here, we highlight how intron retention and, more broadly, alternative splicing coupled to nonsense-mediated mRNA decay (AS-NMD) can be used to regulate gene expression and how this is deregulated in disease. We suggest that the importance of AS-NMD is not restricted to the haematopoietic system but that it plays a prominent role in other normal and aberrant biological settings” (Ge and Porse 2014).
- On intron retention, see also introns under Creation of mRNA variants below.
- A rather dramatic finding: “The conventional model for splicing involves excision of each intron in one piece; we demonstrate this inaccurately describes splicing in many human genes. First, after switching on transcription of SAMD4A, a gene with a 134 kb-long first intron, splicing joins the 3′ end of exon 1 to successive points within intron 1 well before the acceptor site at exon 2 is made. Second, genome-wide analysis shows that >60% of active genes yield products generated by such intermediate intron splicing. These products are present at ∼15% the levels of primary transcripts, are encoded by conserved sequences similar to those found at canonical acceptors, and marked by distinctive structural and epigenetic features. Finally, using targeted genome editing, we demonstrate that inhibiting the formation of these splicing intermediates affects efficient exon–exon splicing. These findings greatly expand the functional and regulatory complexity of the human transcriptome” (Kelly, Georgomanolis, Zirkel et al. 2015, doi:10.1093/nar/gkv386).
- In Arabidopsis (mustard plant), “we found unusual AS [alternative splicing] events inside annotated protein-coding exons. Here, we also identify such AS events in human and use these two sets to analyse their features, regulation, functional impact, and evolutionary origin. As these events involve introns with features of both introns and protein-coding exons, we name them exitrons (exonic introns). [Splicing of exitrons] results in transcripts with different fates. About half of the 1002 Arabidopsis and 923 human exitrons have sizes of multiples of 3 nucleotides (nt). Splicing of these exitrons results in internally deleted proteins and affects protein domains, disordered regions, and various post-translational modification sites, thus broadly impacting protein function. Exitron splicing is regulated across tissues, in response to stress and in carcinogenesis. Intriguingly, annotated intronless genes can be also alternatively spliced via exitron usage ... Altogether, our studies show that exitron splicing is a conserved strategy for increasing proteome plasticity in plants and animals, complementing the repertoire of AS events” (Marquez, Höpfler, Ayatollahi et al. 2015, doi:10.1101/gr.186585.114).
- It’s not only that, as described above, different isoforms produced by splicing can have differing or opposing (repressive or activating) influences on gene expression. A given factor involved in the splicing process itself can play different, context-specific roles in splicing. “Many splicing factors have the ability to behave as splicing repressors for some alternative cassette exons and as splicing activators for others. Unexpectedly, we found that the ability of a given alternative splicing factor to behave as an enhancer or repressor of a specific splicing event can change during development” (Barberan-Soler, Medina, Estella et al. 2010).
- More broadly, “Nearly all ‘activators’ of splicing can, in some cases, function as repressors, and nearly all ‘repressors’ have been shown to function as activators...it is clear that context affects function” (Nilsen and Graveley 2010). For example, “SR [serine/arginine-rich] proteins enhance splicing only when they are recruited to the exon. However, they interfere with splicing by simply relocating them to the opposite intronic side of the splice site”. Other splicing factors (heterogeneous ribonucleoproteins) also can have opposite effects, but the rule of their behavior is the reverse of the one for the SR proteins (Erkelenz, Mueller, Evans et al. 2013).
- There are several classes of regulatory sequences within genes themselves that play a critical role in splicing. One such class consists of “intronic splicing enhancers” (ISEs). A survey of a limited number of these sequences in one human cell line turned up more than a hundred ISEs. “A single ISE element can be bound by multiple factors with distinct activities, and the same factor can recognize multiple ISEs, which suggests that a complicated web of RNA-protein interactions controls splicing to achieve a certain degree of regulatory plasticity” (Wang, Ma, Xiao and Wang 2012). As is usual in such cases, the authors proceed to refer to the challenge of understanding “the splicing code”.
- Adenosine-to-inosine editing of pre-mRNA by ADAR [adenosine deaminases acting on RNA] enzymes “was found to affect splicing regulatory elements within exons. Cassette exons [that is, exons optionally incorporated into mRNA by alternative splicing] were found to be significantly enriched with A-to-I RNA editing sites compared with constitutive exons. ... ADAR knockdown in hepatocarcinoma and myelogenous leukemia cell lines leads to global changes in gene expression, with hundreds of genes changing their splicing patterns in both cell lines. ... Genes showing significant changes in their splicing pattern are frequently involved in RNA processing and splicing activity. ... Our global analysis reveals that ADAR plays a major role in splicing regulation. Although direct editing of the splicing motifs does occur, we suggest it is not likely to be the primary mechanism for ADAR-mediated regulation of alternative splicing. Rather, this regulation is achieved by modulating trans-acting factors involved in the splicing machinery” (Solomon, Oren, Safran et al. 2013).
- Introns in 5'- and 3'-UTRs: Introns within the 5' and 3' untranslated regions have generally been ignored as functionally insignificant, if only because they couldn’t mediate the formation of different protein isoforms. But this is now changing, as other functions are being discovered. In fact, the very “presence of an intron and the act of its removal by the spliceosome can influence almost every step in gene expression from transcription and polyadenylation to mRNA export, localization, translation, and decay”. “All introns can influence gene expression regardless of their position relative to the coding region because they alter the protein makeup of the mRNA protein particle” and this in turn is involved in many aspects of gene expression regulation (Bicknell, Cenik, Chua et al. 2012).
- Alternative splicing and intrinsically unstructured (disordered) proteins: Proteins with unstructured regions tend to play central roles as “hubs” for interaction with many other proteins due to the flexibility of those unstructured regions. Interestingly, “Tissue-specific splicing events appear to alter the disordered regions that harbor binding motifs while leaving the structured regions intact. This can lead to rewiring of molecular interaction networks and new functional consequences” (Babu, Kriwacki and Pappu 2012).
- More on intrinsically disordered proteins as splicing factors: “IDPs have a key role in both constitutive pre-mRNA splicing and alternative splicing ... The protein components of the spliceosome are highly enriched in intrinsic disorder ... Proteins involved in spliceosome assembly and mRNA recognition, such as the retention and splicing complex RES, have a strong propensity for disorder, whereas proteins like the small nuclear ribonucleoprotein particle proteins that comprise the catalytic core of the spliceosome tend to be highly ordered. Spliceosome assembly and conformational rearrangement is regulated by reversible post-translational modifications in disordered regions. Splicing of pre-mRNA is regulated through a dynamic cycle of multisite phosphorylation and dephosphorylation of Ser residues in intrinsically disordered Arg- and Ser-rich regions (termed RS or SR domains) of splicing factors. Recent NMR studies show that unphosphorylated RS domains are fully disordered and highly dynamic and are susceptible to efficient phosphorylation by a number of kinases. Multisite phosphorylation acts as a dynamic switch that favours a more rigid arch-like structure, with well-defined orientations of the Arg and Ser side chains in the RS repeats. The extent of ordering of the RS domain depends on the number of RS repeats and the number of phosphoryl groups. It has been suggested that the interactions of the RS domain with RNA and with other proteins are modulated through entropic changes and the increased charge associated with progressive phosphorylation. Indeed, recent evidence suggests a role for RS domains in regulating the compartmentalization of splicing factors within the nucleus” (Wright and Dyson 2015, doi:10.1038/nrm3920).
- Splice sites “are highly diverse, considering that thousands of different sequences act as naturally occurring splice sites in the human transcriptome”. This diversity, as much of the above indicates, is not the diversity of abstract code, but of highly articulated form. One aspect of that form has to do with “bulged” nucleotides — nucleotides that, due to their spatial displacement from the canonical form of the mRNA helix, can be skipped over in base pairing with the U1 snRNA splicing factor. It is estimated that about 5% of 5' splice sites in 6577 tested human genes involve bulged nucleotides (Roca, Akerman, Gaus et al. 2012).
- There are various splicing factors other than RNA-binding proteins. For example, A small molecule binding to an RNA riboswitch affects alternative splicing in the fungus Neurospora crassa by inducing changes in pre-mRNA structure” (Witten and Ule 2011).
- Likewise: “Pre-mRNA interactions with noncoding RNAs, including a small nucleolar RNA and an RNA related to 5S ribosomal RNA have also been reported” (Witten and Ule 2011). Luco and Misteli 2011: “ncRNAs [noncoding RNAs] have recently emerged as novel regulators of alternative splicing. One mode of control by ncRNAs [among others discussed] is the regulation of the expression of key splicing factors by short microRNAs during development and differentiation”. A long noncoding RNA is thought to sequester protein splicing factors in nuclear splicing speckles until needed; downregulating the noncoding RNA “leads to enhanced exon inclusion in a number of genes”.
- And again: lipids can play a role. See under “Regulation and integration of the regulators” below.
- DNA methylation and binding of DNA by the CTCF protein (see “Insulator protein CTCF” below) can have mutually antagonistic effects upon splicing. “We provide the first evidence that a DNA-binding protein [CTCF] can promote inclusion of weak upstream exons by mediating local RNA polymerase II pausing. ... We further show that CTCF binding to [a particular exon] is inhibited by DNA methylation. ... These findings provide a mechanistic basis for developmental regulation of splicing outcome through heritable epigenetic marks”. And further: “We predict that our identification of CTCF as a DNA-binding regulator of alternative pre-mRNA splicing represents the tip of the iceberg, and that a long list of location-specific DNA-binding ‘splicing factors’ will follow” (Shukla, Kavak, Gregory et al. 2011).
- Splicing can be regulated by the regulated degradation of protein splicing factors. Alternatively, “some splicing events are controlled by signal-induced changes in the localization or accessibility of crucial regulatory proteins” (Heyd and Lynch 2011).
- Most of the RNA-binding proteins (RBPs) that regulate alternative splicing events in cancer tumors “have pleiotropic [multifaceted] effects on splicing and other processes (especially translation), meaning that the critical changes in alternative splicing come as part of a wider program of RBP-mediated changes in gene expression” (David and Manley 2010).
- “Alternative splicing is an integral part of differentiation and developmental programs and contributes to cell lineage and tissue identity as indicated by the mapping of more than 22,000 tissue-specific alternative transcript events in a recent genome-wide sequencing study of tissue-specific alternative splicing” (Luco, Allo, Schor et al. 2011). During differentiation of a myoblast cell line, there were numerous transitions in alternative splicing and changes in the abundance of splicing regulators, suggesting that alternative splicing is “highly regulated” by multiple factors and plays a “major role” in myogenic differentiation (Bland, Wang, Vu et al. 2010).
- An example of tissue-specific splicing: one team of investigators “identified an alternative splice junction used by nuclear factor I/B (NFIB), a protein that had previously been implicated in regulating lung and nervous system development. The novel NFIB transcript (NFIB-S) is highly expressed in megakaryocytes [large bone marrow cells that produce platelets] and is shorter than the canonical isoform. Contrary to the canonical isoform, NFIB-S cannot interact with its binding partner NFIC. Overexpression of NFIB-S or NFIC, but not of canonical NFIB, stimulates megakaryocyte maturation, indicating that the shorter isoform is required for this process”. As part of their study, the researchers reported 29,736 previously unannotated splice junctions in several cell lineages derived from human hematopoietic cells (Lokody 2014, doi:10.1038/nrg3847).
- Alternative splicing plays an as yet unidentified role in regulating the stability and degradation of mRNA transcripts, thereby regulating the gene expression that occurs via these transcripts. It’s been found that alternatively spliced mRNAs with the same 3'-untranslated region (the region that has been thought to contain most RNA stabilizing and destabilizing elements) can be differentially degraded in certain cell types (’t Hoen, Hirsch, de Meijer et al. 2010).
- However, alternative splicing can also affect the 3'- and 5'-untranslated regions “and consequently modulate translation, stability or localization of mRNA” (Venables, Tazi and Juge 2012).
- Splicing is now known to be closely coupled with transcription. “One clear mechanism of coupling is local regulation of elongation rates, which influences co-transcriptional splicing by determining the amount of time the nascent RNA substrate is available to splicing factors before 3' end cleavage and release. First, local changes in elongation can be caused by sequence-specific thermodynamic differences in the transcription bubble. Second, nucleosome positioning can influence elongation and co-transcriptional splicing by (i) locally stalling Pol II and/or (ii) providing a local scaffold for recruitment of positive or negative splicing regulators via modified histone tails. Third, specific recruitment of transcription and RNA processing factors to the Pol II holoenzyme and/or CTD [C terminal domain] plays additional roles” (Oesterreich, Bieberstein and Neugebauer 2011).
- While most splicing seems to occur during transcription, it can also occur post-transcriptionally. In the latter case it appears that “some splicing events are regulated by specific developmental cues or external signals long after the completion of transcription” (Han, Xiong, Wang and Fu 2011).
- Recent research has shown a remarkably common role for antisense transcripts (that is, transcripts from the opposite DNA strand) in regulating the alternative splicing of genes. It is not yet known how this regulation is achieved (Morrissy, Griffith and Marra 2011).
- Article title: “The Splicing Landscape Is Globally Reprogrammed during Male Meiosis” (Schmid, Grellscheid, Ehrmann et al. 2013). The authors’ conclusion: “Our data suggest that there are substantial changes in the determinants and patterns of alternative splicing in the mitotic-to-meiotic transition of the germ cell cycle”.
- A splicing event in a single splicing regulator alters the large-scale pattern of splicing: “The functions of species- and lineage-specific splice variants are largely unknown. Here we show that mammalian-specific skipping of polypyrimidine tract–binding protein 1 (PTBP1) exon 9 alters the splicing regulatory activities of PTBP1 and affects the inclusion levels of numerous exons. During neurogenesis, skipping of exon 9 reduces PTBP1 repressive activity so as to facilitate activation of a brain-specific alternative splicing program. Engineered skipping of the orthologous exon in chicken cells induces a large number of mammalian-like alternative splicing changes in PTBP1 target exons (Gueroussov, Gonatopoulos-Pournatzis, Irimia et al. 2015, doi:10.1126/science.aaa8381).
- For just a glimpse of how complex things can get: “The auxiliary factor of U2 small nuclear ribonucleoprotein (U2AF) facilitates branch point (BP) recognition and formation of lariat introns. The gene for the 35-kD subunit of U2AF gives rise to two protein isoforms (termed U2AF35a and U2AF35b) that are encoded by alternatively spliced exons 3 and Ab, respectively. The splicing recognition sequences of exon 3 are less favorable than exon Ab, yet U2AF35a expression is higher than U2AF35b across tissues. We show that U2AF35b repression is facilitated by weak, closely spaced BPs next to a long polypyrimidine tract of exon Ab. Each BP lacked canonical uridines at position -2 relative to the BP adenines, with efficient U2 base-pairing interactions predicted only for shifted registers reminiscent of programmed ribosomal frameshifting. The BP cluster was compensated by interactions involving unpaired cytosines in an upstream, EvoFold-predicted stem loop (termed ESL) that binds FUBP1/2. Exon Ab inclusion correlated with predicted free energies of mutant ESLs, suggesting that the ESL operates as a conserved rheostat between long inverted repeats upstream of each exon. The isoform-specific U2AF35 expression was U2AF65-dependent, required interactions between the U2AF-homology motif (UHM) and the α6 helix of U2AF35, and was fine-tuned by exon Ab/3 variants. Finally, we identify tandem homologous exons regulated by U2AF and show that their preferential responses to U2AF65-related proteins and SRSF3 are associated with unpaired pre-mRNA segments upstream of U2AF-repressed 3' splice site. These results provide new insights into tissue-specific subfunctionalization of duplicated exons in vertebrate evolution and expand the repertoire of exon repression mechanisms that control alternative splicing” (Kralovicova and Vorechovsky 2016, 10.1093/nar/gkw733).
-
With increasingly sophisticated methods for interrogating molecular
activities within cells, information about splicing is becoming ever
more detailed, revealing cell type-specific, cell cycle-specific,
disease-specific, and, in general, every imaginable sort of
context-specific regulation of splicing. Perhaps the best way to get
a feel for this is simply to consider the titles of a few of the
articles now appearing — and then multiply what you see by 10,000,
with ramifications in every direction of molecular biological
investigation:
● “TDP-43 Affects Splicing Profiles and Isoform Production of Genes Involved in the Apoptotic and Mitotic Cellular Pathways” (De Conti, Akinyi, Mendoza-Maldonado et al. 2015, doi:10.1093/nar/gkv814).
● “TRAP150 Interacts with the RNA-Binding Domain of PSF and Antagonizes Splicing of Numerous PSF-Target Genes in T Cells” (Yarosh, Tapescu, Thompson et al. 2015, doi:10.1093/nar/gkv816).
● “The DNA Replication Licensing Factor Miniature Chromosome Maintenance 7 Is Essential for RNA Splicing of Epidermal Growth Factor Receptor, c-Met, and Platelet-derived Growth Factor Receptor” (Chen, Yu, Michalopoulos et al. 2015, doi:10.1074/jbc.M114.622761).
● “The DNA Replication Licensing Factor Miniature Chromosome Maintenance 7 is Essential for RNA Splicing of Epidermal Growth Factor Receptor, c-met and Platelet Derived Growth Factor Receptor” (Luo, Chen and Yu 2015, doi:10.1096/fj.1530-6860).
● “Meta-Analysis of Multiple Sclerosis Microarray Data Reveals Dysregulation in RNA Splicing Regulatory Genes” (Paraboschi, Cardamone, Rimoldi et al. 2015, doi:10.3390/ijms161023463).
● “The Alternative Splicing of Cytoplasmic Polyadenylation Element Binding Protein 2 Drives Anoikis Resistance and the Metastasis of Triple Negative Breast Cancer” (Johnson, Vu, Griffin et al. 2015, doi:10.1074/jbc.M115.671206).
● “Arginine Methylation and Citrullination of Splicing Factor Proline- and Glutamine-Rich (SFPQ/PSF) Regulates Its Association with mRNA” (Snijders, Hautbergue, Bloom et al. 2015, doi:10.1261/rna.045138.114).
- “Different RBPs [RNA-binding proteins] regulate splicing during brain development. Among them are polypyrimidine tract-binding protein 1 (PTBP1), PTBP2 and Ser/Arg repetitive matrix protein 4 (SRRM4), levels of which change during neurogenesis. Therefore, alterations of their target splicing networks occur during the transition from neural progenitors to fully differentiated neurons. In particular, PTBP1 and PTBP2 engage in a crosstalk, whereby in neuronal progenitors PTBP1 represses the inclusion of PTBP2 exon 10, leading to exon skipping and a transcript with a premature termination codon and NMD [nonsense-mediated RNA decay]. As cells exit the cell cycle to differentiate into neurons, PTBP1 is downregulated, whereas SRRM4, which acts as a positive regulator of PTBP2 splicing, is upregulated, and this promotes PTBP2 exon 10 inclusion. As a result, PTBP2 is expressed, promoting neuronal development and tissue maintenance” (Baralle and Giudice 2017, doi:10.1038/nrm.2017.27).
- “Another mechanism of splicing regulation by RBPs [RNA-binding proteins] in neuronal differentiation is through their own alternative splicing changes during development. This occurs with RNA-binding protein FOX1 homologue 1 (RBFOX1) — an RBP that has been associated with both neuronal differentiation and neurodevelopmental programmes that control synaptic functions. Exon 19 of RBFOX1 is alternatively spliced by RBFOX proteins, giving rise to nuclear (excluding exon 19) or cytoplasmic (including exon 19) protein isoforms. In RBFOX-depleted neurons, more than 500 alternatively spliced cassette exons were misregulated, leading to significant changes in the level of exon inclusion or skipping in comparison with the control condition. Exogenous introduction of the nuclear isoform rescues these splicing changes, probably by binding to the GCAUG motifs in the proximal intronic region downstream of the regulated exons. By contrast, expression of the cytoplasmic variant rescues changes in mRNA levels of synaptic and autism-related genes through mRNA stabilization mechanisms (3' UTR binding and competition with microRNAs)” (Baralle and Giudice 2017, doi:10.1038/nrm.2017.27).
- “One mechanism that contributes to splicing fidelity is the repression of nonconserved cryptic exons by splicing factors that recognize dinucleotide repeats. We previously identified that TDP-43 and PTBP1/PTBP2 are capable of repressing cryptic exons utilizing UG and CU repeats, respectively. Here we demonstrate that hnRNP L (HNRNPL) also represses cryptic exons by utilizing exonic CA repeats, particularly near the 5’SS. We hypothesize that hnRNP L regulates CA repeat repression for both cryptic exon repression and developmental processes such as T cell differentiation” (McClory, Lynch and Ling 2018, doi:10.1261/rna.065508.117).
- “Alternative splicing (AS) plays important roles in embryonic stem cell (ESC) differentiation. In this study, we first identified transcripts that display specific AS patterns in pluripotent human ESCs (hESCs) relative to differentiated cells. One of these encodes T-cell factor 3 (TCF3), a transcription factor that plays important roles in ESC differentiation. AS creates two TCF3 isoforms, E12 and E47, and we identified two related splicing factors, heterogeneous nuclear ribonucleoproteins (hnRNPs) H1 and F (hnRNP H/F), that regulate TCF3 splicing. We found that hnRNP H/F levels are high in hESCs, leading to high E12 expression, but decrease during differentiation, switching splicing to produce elevated E47 levels. Importantly, hnRNP H/F knockdown not only recapitulated the switch in TCF3 AS but also destabilized hESC colonies and induced differentiation. Providing an explanation for this, we show that expression of known TCF3 target E-cadherin, critical for maintaining ESC pluripotency, is repressed by E47 but not by E12” (Yamazaki, Liu, Lazarev et al. 2018, doi:10.1101/gad.316984.118).
- “We uncovered a noncanonical function of [splicing factor] U2AF1, showing that it directly binds mature mRNA in the cytoplasm and negatively regulates mRNA translation. This splicing-independent role of U2AF1 is altered by the S34F mutation, and ... the mutation affects translation of hundreds of mRNA. One functional consequence is increased synthesis of the secreted chemokine interleukin, which contributes to metastasis, inflammation, and cancer progression in mice and humans” (Palangat, Anastasakis, Fei et al. 2019, doi:10.1101/gad.319590.118).
- “Recent evidence points to a regulatory role of chromatin-related proteins in alternative splicing regulation. Using an unbiased approach, we have identified the acetyltransferase p300 as a key chromatin-related regulator of alternative splicing. p300 promotes genome-wide exon inclusion in both a transcription-dependent and -independent manner. Using CD44 as a paradigm, we found that p300 regulates alternative splicing by modulating the binding of splicing factors to pre-mRNA. Using a tethering strategy, we found that binding of p300 to the CD44 promoter region promotes CD44v exon inclusion independently of RNAPII transcriptional elongation rate. Promoter-bound p300 regulates alternative splicing by acetylating splicing factors, leading to exclusion of hnRNP M from CD44 pre-mRNA and activation of Sam68. p300-mediated CD44 alternative splicing reduces cell motility and promotes epithelial features” (Siam, Baker, Amit et al., doi:10.1261/rna.069856.118).
- “TCF3, also known as E2A, is a well-studied transcription factor that plays an important role in stem cell maintenance and hematopoietic development. The TCF3 gene encodes two related proteins, E12 and E47, which arise from mutually exclusive alternative splicing (MEAS). Since these two proteins have different DNA binding and dimerization domains, this AS event must be strictly regulated to ensure proper isoform ratios. Previously, we found that heterogeneous nuclear ribonucleoprotein (hnRNP) H1/F regulates TCF3 AS by binding to exonic splicing silencers (ESSs) in exon 18b. Here, we identify conserved intronic splicing silencers (ISSs) located between, and far from, the two mutually exclusive exons, and show that they are essential for MEAS. Further, we demonstrate that the hnRNP PTBP1 binds the ISS and is a regulator of TCF3 AS. We also demonstrate that hnRNP H1 and PTBP1 regulate TCF3 AS reciprocally, and that position-dependent interactions between these factors are essential for proper TCF3 MEAS. Our study provides a new model in which MEAS is regulated by cooperative actions of distinct hnRNPs bound to ISSs and ESSs” (Yamazaki, Liu and Manley 2019, doi:10.1261/rna.072298.119).
- “A critical role for alternative pre-mRNA processing in cell migration has emerged in axon outgrowth during neuronal development, immune cell migration, and cancer metastasis. These findings suggest that migratory signals result in expression changes of post-translational modifications of splicing or polyadenylation factors, leading to splicing events that generate promigratory isoforms” (TOC blurb for Mitra, Lee and Coller 2019, doi:10.1016/j.tcb.2019.10.007).
- “Alternative splicing generates distinct mRNA variants and is essential for development, homeostasis, and renewal. Proteins of the serine/arginine (SR)-rich splicing factor family are major splicing regulators that are broadly required for organ development as well as cell and organism viability ... Here, we used the continuously growing mouse incisor as a model to dissect the functions of the prototypical SR family protein SRSF1 during tissue homeostasis and renewal. We identified an SRSF1-governed alternative splicing network that is specifically required for dental proliferation and survival of progenitors [that is, progenitor cells] but dispensable for the viability of differentiated cells. We also observed a similar progenitor-specific role of SRSF1 in the small intestinal epithelium, indicating a conserved function of SRSF1 across adult epithelial tissues. Thus, our findings define a regulatory mechanism by which SRSF1 specifically controls progenitor-specific alternative splicing events to support adult tissue homeostasis and renewal” (Yu, Cazares, Tang et al. 2022, doi:10.1016/j.devcel.2022.01.011).
-
Microexon splicing
“Microexons, defined here as 3-27 nucleotide (nt)-long exons, have
been largely missed” in alternative splicing and related studies.
“This is especially true for microexons shorter than 15 nt”
(Irimia, Weatheritt, Ellis et al. 2014).
- “Here, we define the largest program of functionally coordinated, neural-regulated AS [alternative splicing] described to date in mammals. Relative to all other types of AS within this program, 3-15 nucleotide “microexons” display the most striking evolutionary conservation and switch-like regulation. [The proteins expressed from] these microexons modulate the function of interaction domains of proteins involved in neurogenesis. Most neural microexons are regulated by the neuronal-specific splicing factor nSR100/SRRM4, through its binding to adjacent intronic enhancer motifs. Neural microexons are frequently misregulated in the brains of individuals with autism spectrum disorder, and this misregulation is associated with reduced levels of nSR100. The results thus reveal a highly conserved program of dynamic microexon regulation associated with the remodeling of protein-interaction networks during neurogenesis, the misregulation of which is linked to autism” (Irimia, Weatheritt, Ellis et al. 2014).
-
tRNA splicing
- Two studies on a novel syndrome of the central and peripheral nervous systems “implicate defective tRNA splicing as the underlying molecular cause of the syndrome, thus adding to a growing body of literature that links neurological diseases with tRNA modifications” (Koch 2014).
-
Role of the minor spliceosome
The foregoing references to the “spliceosome” (the complex of small
RNAs and proteins that carry out the splicing operations) generally
pertain to the “major” splicesome. There is a “minor” splicesome
that doesn’t get as much press, because it typically comes to bear
on only one of possibly many introns in a given pre-mRNA — and this
only in the case of several hundred from among our 20,000 or so
protein-coding genes.
- One component of the minor spliceosome (the ribonucleoprotein U6atac) has been found to be “strikingly unstable” under usual conditions, and its scarcity means that the failure to splice a single minor intron in a pre-mRNA may delay or prevent its translation into protein, even if all the major introns have already been spliced. But, under stress, a signaling enzyme stabilizes U6atac, allowing the minor intron to be spliced. This can result in a sudden and dramatic increase in translation levels of minor intron-containing mRNAs. In the other direction, a reduction in transcription of U6atac itself can rapidly reduce the pool of such molecules in the cell, due to their instability. “Thus, minor introns function as control switches that are embedded in hundreds of genes and regulated by U6atac abundance” (Younis, Dittmar, Wang et al. 2013). The regulation of minor-intron splicing can have large effects on gene expression relating to cell growth and differentiation, inflammatory response, and tumor formation, among other things.
-
Role of nuclear organization
- “Regulation of the availability of splicing components provides a potentially powerful means by which constitutive and alternative splicing events may be controlled. The highly compartmentalized nature of the cell nucleus, which contains several different types of nonmembranous substructures, or ‘bodies,’ that concentrate RNA processing factors, provides such a regulatory architecture. Among the domains that concentrate splicing and other RNA processing factors are inter-chromatin granule clusters or ‘speckles,’ paraspeckles, Cajal Bodies and nuclear stress bodies” (Braunschweig, Gueroussov, Plocik et al. 2013).
- “How the nuclear machinery executes a high-precision operation such as splicing over genomic distances that may exceed 1 Mb is currently unknown. The most straightforward explanation is that, analogous to enhancers and their target promoters, these transcript components are physically approximated to one another through direct chromatin interactions” (Stamatoyannopoulos 2012) — which, of course, only pushes the problem back one step, since now there is the question how the right genomic sequences are brought into proximity (Braunschweig, Gueroussov, Plocik et al. 2013).
- “Splicing factors can shuttle between speckles and nearby sites of nascent RNA transcription, and ... this shuttling behavior can be controlled by specific kinases and phosphatases that alter the posttranslational modification status of SR [serine/arginine-rich] proteins and other splicing factors” (Braunschweig, Gueroussov, Plocik et al. 2013).
- More recent studies have shown a more complex picture, with “spliceosomes localized to regions of decompacted chromatin at the periphery of — or within — nuclear speckles. ... Post-transcriptional splicing occurs in nuclear speckles [consistent with earlier studies which] suggested that the introns of specific transcripts are spliced within speckles” (Braunschweig, Gueroussov, Plocik et al. 2013).
- “Mammalian nuclei typically contain several Cajal bodies, and these domains are thought to represent primary sites of spliceosomal and nonspliceosomal snRNP [protein/small-RNA complex] biogenesis, maturation, and recycling. The formation and size of Cajal bodies relates to the transcriptional and metabolic activity of cells, and these structures are prominent in rapidly proliferating cells” (Braunschweig, Gueroussov, Plocik et al. 2013).
- “Nuclear stress bodies are structures that form specifically in response to a variety of stress conditions including heat shock, oxidative stress, or exposure to toxic materials. These structures are thought to mediate global changes in gene expression, in part by sequestering splicing factors” (Braunschweig, Gueroussov, Plocik et al. 2013).
-
Role of RNA polymerase
“The prevailing view is that most pre-mRNA splicing occurs
co-transcriptionally when the nascent transcript is still attached
to the DNA by RNA polymerase II, and adjacent exons are spliced
before the rest of the gene is transcribed” (de Almeida and
Carmo-Fonseca 2012). So much of the discussion of alternative
splicing could have been included under
DECISION-MAKING DURING TRANSCRIPTION
above.
- Histone modifications that slow down the rate of transcription elongation by RNA polymerase can lead to the inclusion of exons by splicing, whereas modifications that tend toward the formation of open (“relaxed”) chromatin and encourage a rapid rate of elongation can lead to the exclusion of exons. The assumption is that “slowing down the elongating polymerase facilitates assembly of the spliceosome at suboptimal splice sites of alternative exons” (de Almeida and Carmo-Fonseca 2012).
- For example, elevated levels of H3K9 trimethylation (along with heterochromatin protein 1 — HP1γ) were found in one study to be “characteristic of several genes”. HP1γ “facilitates inclusion of the alternative exons via a mechanism involving decreased RNA polymerase II elongation rate” (Saint-André, Violaine, Eric Batsché, Christophe Rachez and Christian Murchardt 2011).
- RNA polymerase elongation can also be slowed or paused by DNA-binding proteins that bind to the region of alternative exons. “Indeed...the DNA-binding protein CTCF binds intragenically, causes local RNA polymerase II pausing and stimulates inclusion of weak upstream exons” (de Almeida and Carmo-Fonseca 2012).
- It’s not only that the transcribing enzyme plays a role in splicing regulation. Splicing processes can in turn regulate transcription. “Introns have a stimulatory effect on gene expression in both yeast and mice, and a growing body of recent evidence indicates that the mechanism involves a direct effect of splicing on initiation, elongation and termination of RNAP II-dependent transcription”. Certain splicing factors interact with an elongation factor in vitro, stimulating transcription “in a manner that is dependent on the presence of functional splice sites in the pre-mRNA”. In vivo, depletion of certain splicing factors “triggered a widespread defect in transcription elongation” (de Almeida and Carmo-Fonseca 2012).
- “Other studies showed that in addition to transcriptional elongation, splicing can also stimulate transcription initiation both in vitro and in vivo”. And splicing is also linked to transcription termination, so that “splicing appears to feed back to RNAP II during all stages of transcription” (de Almeida and Carmo-Fonseca 2012).
- “Interactions between the splicing machinery and RNA polymerase II increase protein-coding gene transcription. Similarly, exons and splicing signals of enhancer-generated long noncoding RNAs (elncRNAs) augment enhancer activity. However, elncRNAs are inefficiently spliced, suggesting that, compared with protein-coding genes, they contain qualitatively different exons with a limited ability to drive splicing. We show here that the inefficiently spliced first exons of elncRNAs as well as promoter-antisense long noncoding RNAs (pa-lncRNAs) in human and mouse cells trigger a transcription termination checkpoint that requires WDR82, an RNA polymerase II–binding protein, and its RNA-binding partner of previously unknown function, ZC3H4. We propose that the first exons of elncRNAs and pa-lncRNAs are an intrinsic component of a regulatory mechanism that, on the one hand, maximizes the activity of these cis-regulatory elements by recruiting the splicing machinery and, on the other, contains elements that suppress pervasive extragenic transcription” (Austenaa, Piccolo, Russo et al. 2021, doi:10.1038/s41594-021-00572-y).
-
Role of RNA secondary and tertiary structure
“Structured mRNA regions can affect alternative splicing regulation
at different levels, including the availability of
cis-regulatory sequences, interaction of splicing factors,
and variations in the critical distances between binding motifs”
(Wachter 2014).
- An RNA-protein complex that catalyzes the removal of introns recognizes the 5' end of the intron to be removed. It has been unclear how it recognizes the other (3' or “downstream”) end. It now appears that the secondary structure (the folding at a certain stage) of the RNA being spliced plays a role. Meyer et al. (2011) found that for many introns in yeast this folding brought one or more 3' splice sites within reach of the RNA-protein splicing complex, whereupon the complex could utilize any one of the splice sites (consisting of short, two- or three-letter sequences) that was neither too close nor too far from the splicing complex. Furthermore, in the case of one gene they investigated, the thermal stability of the RNA’s secondary structure influenced which 3' splice site was chosen; a temperature change could alter the choice. It all points to a regulatory role in splicing for the RNA folding structure.
-
There are actually many different ways the secondary and
tertiary structure of RNAs affect splicing (briefly reviewed
by McManus and Graveley 2011). For example:
- “There are many examples of local pre-mRNA structures that regulate alternative splicing, often by preventing spliceosomal recognition of the 5' splice site, 3' splice site, and branch point sequence elements”.
- Regulatory sequences in the mRNA that recruit other regulatory elements can have greater or lesser effects depending on whether they are sequestered in RNA structures.
- Long-range structures in pre-mRNAs can also play a role. For example, in the Drosophila gene from which some 38,000 isoforms can be derived, there are “docking sequences” and various “selector sequences” that can base pair with the docking sequences. The distances between the two types of sequence can be considerable, and they must be brought together by means of appropriate folding of the RNA.
- There are various different sorts of interaction between RNA folding structures and splicing regulatory proteins — interactions that have a direct bearing on the splicing results.
- “RNA structures can also change in response to binding small molecules, and this may be an important mechanism of splicing regulation”.
- The RNA structures investigated for their effects upon splicing have generally been short-range structures. But now, given a method to detect both local and long-range structures with equal effectiveness, researchers report that “long-range base-pairings carry an important, yet unconsidered part of the splicing code, and...even by modest estimates, there must be thousands of such potentially regulatory structures conserved throughout the evolutionary history of mammals”. “We estimate that splicing of thousands of mammalian genes is dependent on RNA structures, including ones which act over long ranges” (Pervouchine, Khrameeva, Pichugina et al. 2011).
- Drosha regulates Drosha — via a hairpin structure: “Apart from its central role in the biogenesis of miRNAs, DROSHA is also known to recognize and cleave miRNA-like hairpins in a subset of transcripts without apparent small RNA production. Here, we report that the human DROSHA transcript is one such noncanonical target of DROSHA. Mammalian DROSHA genes have evolved a conserved hairpin structure spanning a specific exon–intron junction, which serves as a substrate for the Microprocessor in human cells but not in murine cells. We show that it is this hairpin element that decides whether the overlapping exon is alternatively or constitutively spliced. We further demonstrate that DROSHA promotes skipping of the overlapping exon in human cells independently of its cleavage function. Our findings add to the expanding list of noncanonical DROSHA functions” (Lee, Nam and Shin 2017, doi:10.1261/rna.059808.116).
- Regarding the choice of aberrant, disease-causing splice sites in the LMNA RNA: “While splice site choice is in part defined by sequence complementarity to U1 snRNA, we identify RNA secondary structural elements near the alternative 5′ splice sites and show that splice site choice is significantly influenced by the structural context of the available splice sites. Furthermore, relative positioning of the competing sites within the primary sequence of the pre-mRNA is a predictor of 5′ splice site usage, with the distal position favored over the proximal, regardless of sequence composition. Together, these results demonstrate that 5′ splice site selection in LMNA is determined by an intricate interplay among RNA sequence, secondary structure and splice site position” (Shilo, Tosto, Rausch et al. 2019, doi:10.1093/nar/gkz259).
-
Role of temperature
- Mammalian circadian rhythms are interwoven with daily rhythms in body temperature, and the cold-inducible RNA-binding protein, CIRBP, helps to maintain these temperature rhythms. In a study of mouse fibroblasts, researchers have found that a modest drop in body temperature (from 38°C to 33°C) resulted in a remarkably high increase in expression of the Cirbp mRNA. This increase turned out to be due to a temperature-dependent change in the splicing of Cirbp; splicing became much more efficient at the cooler temperature. A 337-base-pair region within intron 1 of the mRNA was shown to be sufficient for conferring the temperature sensitivity — apparently by means still unknown. Also, the work suggested that “Cirbp is not the only mRNA regulated by this mechanism and that subtle changes in temperature likely regulate many other mRNAs through gene-specific changes in splicing efficiency” (Green 2016, doi:10.1101/gad.289587.116).
- Evidence from many different mRNA splicing events “suggests that body temperature changes are sufficient for the regulation of alternative splicing in vivo [and] that alternative splicing acts like a thermometer to sense body temperature changes, translating these into molecular consequences” (Koch 2017, doi:10.1038/nrg.2017.61).
-
Role of histone modifications and chromatin structure
There are, as so often happens, causal arrows in both directions:
evidence suggests both that “chromatin structure determines
splicing choices, and...splicing can also act as a determinant of
histone modification” (de Almeida and Carmo-Fonseca 2012).
- “Analysis of alternative splicing regulation has traditionally focused on RNA sequence elements and their associated splicing factors, but recent provocative studies point to a key function of chromatin structure and histone modifications in alternative splicing regulation. These insights suggest that epigenetic regulation determines not only what parts of the genome are expressed but also how they are spliced” (Luco, Allo, Schor et al. 2011).
- Proteins that interact with specific histone modifications have been shown to play a role in recruiting splicing factors (de Almeida and Carmo-Fonseca 2012).
- It is proposed that proteins simultaneously play a dual role in splicing regulation: both by binding to DNA and slowing down RNA polymerase elongation (see “Role of RNA polymerase” above) and by recruiting splicing factors. “By having two pathways to transmit a regulatory signal to the output, this circuit [sic] may reject transient activation signals and respond only to persistent signals” (de Almeida and Carmo-Fonseca 2012).
- Splicing activity in turn can affect histone modifications. “H3K36me3 marking is directly influenced by splicing”, probably by enhancing the recruitment of a methylating enzyme to elongating RNA polymerase (de Almeida and Carmo-Fonseca 2012).
- A recent study “provides independent evidence that splicing plays an active role in modulating chromatin structure...Hu proteins, a family of mammalian RNA-binding proteins that participate in splicing regulation through interaction with the spliceosome, can induce local histone hyperacetylation in regions surrounding alternative exons. Hu proteins are recruited to their target binding sites in the pre-mRNA and directly interact with histone deacetylase 2, inhibiting its activity. Consequently, chromatin remains hyperacetylated after passage of the polymerase in a pioneer round of transcription; this in turn increases the local elongation rate of later incoming polymerases, leading to decreased exon inclusion” (de Almeida and Carmo-Fonseca 2012).
-
Role of mitochondria
- “Since eukaryotic gene expression is an energy demanding process, differences in the energy budget of each cell could determine gene expression differences ... We find that changes in mitochondrial content can account for ∼50% of the variability observed in protein levels. This is the combined result of the effect of mitochondria dosage on transcription and translation apparatus content and activities. Moreover, we find that mitochondrial levels have a large impact on alternative splicing, thus modulating both the abundance and type of mRNAs ... The results of this study show that mitochondrial content (and/or probably function) influences mRNA abundance, translation, and alternative splicing, which ultimately affects cellular phenotype” (Guantes, Rastrojo, Neves et al. 2015, doi:10.1101/gr.178426.114).
- “The amount of energy that mitochondria make available for gene expression varies considerably. It depends on: the energetic demands of the tissue; the mitochondrial DNA mutant load; the number of mitochondria; stressors present in the cell. Hence, when failing mitochondria place the cell in energy crisis there are major effects on gene expression affecting the risk of degenerative diseases, cancer and ageing” (Muir, Diot and Poulton 2016, doi:10.1002/bies.201500105).
-
Regulation and integration of the regulators
Distinguishing regulators from what they regulate is always a
rather artificial exercise in the organism. The heading of this
subsection is therefore problematic.
“In addition to generating vast repertoires of RNAs and proteins,
splicing has a profound impact on other gene regulatory layers,
including mRNA transcription, turnover, transport, and translation.
Conversely, factors regulating chromatin and transcription
complexes impact the splicing process. This extensive crosstalk
between gene regulatory layers takes advantage of dynamic spatial,
physical, and temporal organizational properties of the cell
nucleus, and further emphasizes the importance of developing a
multidimensional understanding of splicing control” (Braunschweig,
Gueroussov, Plocik et al. 2013).
“A relatively small number of [alternative-splicing-] regulated
exons can act to rewire entire programs of gene regulation by
modifying core domains of proteins that dictate the activities of
regulators of chromatin, transcription, and other steps in gene
regulation. Numerous other alternative splicing events remodel
protein interaction and signaling networks that are important for
establishing cell type-specific functions. Such alternative
splicing events are often found in disordered domains of proteins
that are subject to phosphorylation and other types of
posttranslational modifications. Interestingly, these domains are
often found in splicing factors and other nuclear gene expression
regulators” (Braunschweig, Gueroussov, Plocik et al. 2013) — which
is to say that alternative splicing often occurs in the regulation
of alternative splicing.
- Here’s a picture of some of the interwoven complexity of RNA splicing: “Splice site selection depends on multiple parameters including the presence of splicing regulators, the strength of splice sites, the structure of exon–intron junctions, and the process of transcription ... Next to conserved cis elements such as the splice donor and acceptor sites, branch sites, polypyrimidine tracts, and a range of other sequence motifs are recognized by various auxiliary splicing factors. These auxiliary RNA-binding proteins (RBPs) are not part of the spliceosomal machinery but can enhance or suppress alternative splicing by interfering with it ... studies have shown that RBPs recognize short (3–7 nucleotides) degenerate motifs, have multiple RNA-binding domains, and display variable efficiency when multiple motifs cluster together. Moreover, many RBPs regulate the expression of other auxiliary factors ... Alternative splicing can also be regulated in a manner totally independent of auxiliary splicing factors. Splicing silencer sequences regulate alternative splicing when competing 5' splice sites are present in the same RNA molecule. The competing 5' splice sites are equally well recognized by the U1 small nuclear ribonucleoprotein (snRNP), but silencer sequences alter the configuration in which U1 binds to the 5' splice sites, leading to silencing of the 5' splice site. This can change the efficiency of a splice site: weak 5' splice sites can be recognized and used instead of stronger 5' splice sites” (Klerk and ’t Hoen 2015, doi:10.1016/j.tig.2015.01.001).
- The U1 RNA is one of several small nuclear RNAs (snRNAs) crucially involved in splicing. The numerous variant copies of U1 snRNA genes in the human genome have long been thought to be pseudogenes (see “Pseudogenes” below). However, many of them produce fully processed transcripts, and an investigation of one of them showed that it “regulates expression of a subset of target genes at the level of pre-mRNA processing”. Furthermore, many of the variant U1 genes are differentially expressed in different cell types, “suggesting developmental control of RNA processing through expression of different sets of vUI snRNPs [variant U1 small nuclear ribonucleoproteins]” (O'Reilly, Dienstbier, Cowley et al. 2013).
- Another layer of regulation: “Splicing regulatory proteins are subject to modification by phosphorylation, acetylation, methylation, sumoylation and hydroxylation”. For example, each of three kinase families phosphorylates certain splicing proteins in distinct ways, “with differing functional consequences” (Heyd and Lynch 2011).
- “Our results indicate that lipids can influence pre-mRNA processing [splicing] by regulating the phosphorylation status of specific regulatory factors, which is mediated by protein phosphatase activity” (Sumanasekera, Kelemen, Beullens et al. 2012).
- The question of integration: “We need to understand how many divergent mechanistic pathways are triggered by a single stimulus. For example, T cell signaling induces [a particular] regulatory program [described in the paper] and it activates at least two other splicing regulatory mechanisms that regulate a non-overlapping set of exons. Similarly, DNA damage triggers multiple splicing-relevant pathways” (Heyd and Lynch 2011).
- “In many aspects, alternative splicing decisions are analogous to transcriptional initiation; multiple factors, both positive and negative, assemble onto a nucleic acid control region, and the combination of assembled factors leads to an integrated decision” (Barberan-Soler, Medina, Estella et al. 2010). Splicing factors bind to the pre-mRNA in a “highly ordered” way, but the binding of individual factors is reversible. This has “important implications for the regulation of alternative splicing": “If spliceosome [a complex of protein splicing factors] assembly is reversible and no single assembly step irreversibly commits a particular pair of splice sites to splicing, then alternative splice site choice can potentially be regulated at any stage of assembly” (Hoskins, Friedman, Gallagher et al. 2011).
- The presence of the structural protein CTCF at a gene tends to cause RNA polymerase II pausing, which in turn promotes the inclusion via splicing of weak upstream exons. On the other hand, DNA methylation in gene bodies — the presence of 5-methylcytosine — promotes exon exclusion by evicting CTCF. Now it is found that the TET1 and TET2 proteins, which can oxidize 5-methylcytosine to 5-hydroxymethylcytosine and 5-carboxylcytosine, help mediate between these two possibilities. When TET proteins reduce DNA methylation by oxidizing 5-methylcytosine at CTCF binding sites in the CD45 gene, the presence of CTCF is encouraged and alternative exon inclusion is facilitated. When TET levels are reduced, resulting in increased DNA methylation, the result is CTCF eviction and exon exclusion. “We further show genomewide that reciprocal exchange of 5‐hydroxymethylcytosine and 5‐methylcytosine at downstream CTCF‐binding sites is a general feature of alternative splicing in naïve and activated CD4+ T cells. These findings significantly expand our current concept of the pre‐mRNA ‘splicing code’ to include dynamic intragenic DNA methylation catalyzed by the TET proteins” (Marina, Sturgill, Bailly et al. 2016, doi:10.15252/embj.201593235).
- In general: RNA-binding proteins and other molecules act cooperatively or competitively in complex fashion to regulate splicing, and other variables contribute to the regulation. “To understand such integrated regulation, RNA splicing maps will need to be combined with analyses of other variables that contribute to alternative splicing decisions, such as splicing kinetics, transcriptional elongation speed, chromatin, the post-translational modifications of RNA-binding proteins, RNA structure and the interactions of pre-mRNA with other noncoding RNAs” (Witten and Ule 2011).
- “Proteins of the Rbfox family act with a complex of proteins called the Large Assembly of Splicing Regulators (LASR). We find that Rbfox interacts with LASR via its C-terminal domain (CTD), and this domain is essential for its splicing activity. In addition to LASR recruitment, a low-complexity sequence within the CTD contains repeated tyrosines that mediate higher-order assembly of Rbfox/LASR and are required for splicing activation by Rbfox ... We find that assembly of the Rbfox CTD plays an essential role in its normal splicing function. Rather than simple recruitment of individual regulators to a target exon, alternative splicing choices also depend on the higher-order assembly of these regulators within the nucleus” (Ying, Wang, Vuong et al. 2017, doi:10.1016/j.cell.2017.06.022).
-
Trans-splicing
Trans-splicing occurs when exons from completely different mRNA
molecules are spliced together. This results in proteins that cannot
at all be said to be directly coded for by any particular gene. Due to
the technical difficulty of detecting trans-splicing events reliably,
not much has been known about their significance. This, however, may
be about to change.
- “We successfully identified and confirmed four trans-spliced RNAs, including the first reported trans-spliced large intergenic noncoding RNA (‘tsRMST’). We showed that these trans-spliced RNAs were all highly expressed in human pluripotent stem cells and differentially expressed during hESC [human embryonic stem cell] differentiation. Our results further indicated that tsRMST can contribute to pluripotency maintenance of hESCs by suppressing lineage-specific gene expression through the recruitment of NANOG and the PRC2 complex factor, SUZ12” (Wu, Yu, Chuang et al. 2014a).
-
Exon shuffling
“In normal splicing, the retained exons of an RNA transcript remain in
the same order as in the DNA template. However, in a recent
development whose significance has yet to be explored, it’s been shown
that some transcripts in humans and other organsms have the order of
their exons rearranged. According to a paper on the topic, “We show
that most PTES (post-transcriptional exon shuffling) transcripts are
expressed in a wide variety of human tissues, that they can be
polyadenylated, and that some are conserved in mouse...[The research]
suggests both that the phenomenon is much more widespread than
previously thought and that some PTES transcripts could be functional”
“Al-Balool, Weber, Liu et al. 2011).
-
Circular RNAs
Circular RNAs can act as microRNAs, serve as protein sponges, regulate
protein functions, and play a role in cap-independent translation, among
other things.
“Thousands of loci in the human and mouse genomes give rise to circular
RNA transcripts; at many of these loci, the predominant RNA isoform is
a circle. Using an improved computational approach for circular RNA
identification, we found widespread circular RNA expression in
Drosophila melanogaster and estimate that in humans, circular RNA may
account for 1% as many molecules as poly(A) RNA. Analysis of data from
the ENCODE consortium revealed that the repertoire of genes expressing
circular RNA, the ratio of circular to linear transcripts for each
gene, and even the pattern of splice isoforms of circular RNAs from
each gene were cell-type specific. These results suggest that
biogenesis of circular RNA is an integral, conserved, and regulated
feature of the gene expression program” (Salzman et al. 2013).
Circular RNAs were once dismissed as genetic accidents or experimental
artifacts. Now, it appears, “the predominance of linear RNAs may have
been the artifact”. At least some of the circular RNAs “act as
molecular ‘sponges’, binding to and blocking ... microRNAs. But the
researchers suspect that the circular RNAs have many other functions.
The molecules comprise ‘a hidden, parallel universe’ of unexplored
RNAs”, says one researcher. Thousands of these new RNAs have been
found in mammals. “‘It’s yet another terrific example of an important
RNA that has flown under the radar’ [according to another researcher].
‘You just wonder when these surprises are going to stop’”. Circular
RNAs “are so abundant, there are probably a multitude of functional
roles”, according to a third researcher (Ledford 2013).
“It is now clear that there is a diversity of circular RNAs in
biological systems. Circular RNAs can be produced by the direct
ligation of 5' and 3' ends of linear RNAs, as intermediates in RNA
processing reactions, or by “backsplicing,” wherein a downstream 5'
splice site (splice donor) is joined to an upstream 3' splice site
(splice acceptor). Circular RNAs have unique properties including the
potential for rolling circle amplification of RNA, the ability to
rearrange the order of genomic information, protection from
exonucleases, and constraints on RNA folding. Circular RNAs can
function as templates for viroid and viral replication, as
intermediates in RNA processing reactions, as regulators of
transcription in cis, as snoRNAs, and as miRNA sponges” (Lasda and
Parker 2014, doi:10.1261/rna.047126.114).
“The identification of EIciRNAs [exon-intron circRNAs] in this study,
together with circRNAs formed exclusively with either exonic or intronic
sequences suggests that there are at least three distinct circRNA
populations in animal cells. Also, certain exonic sequences, which have
been classically viewed as ‘protein-coding’ sequences, contribute to the
formation of at least two types of ‘noncoding’ circular transcripts of
exonic circRNAs and EIciRNAs. It is also fascinating that exon-only
circRNAs may be involved in regulatory functions in the cytoplasm,
whereas the EIciRNAs identified in this study appear to be efficiently
retained for transcriptional regulation in the nucleus. Furthermore, we
speculate that the functions and related mechanisms of circRNAs may be
rather diverse” (Li, Huang, Bao et al. 2015, doi:10.1038/nsmb.2959).
“We present a comprehensive investigation of circRNA expression profiles
across 11 tissues and four developmental stages in rats, along with
cross-species analyses in humans and mice. Although the expression of
circRNAs is positively correlated with that of cognate mRNAs, highly
expressed genes tend to splice a larger fraction of circular transcripts.
Moreover, circRNAs exhibit higher tissue specificity than cognate mRNAs.
Intriguingly, while we observed a monotonic increase of circRNA abundance
with age in the rat brain, we further discovered a dynamic, age-dependent
pattern of circRNA expression in the testes that is characterized by a
dramatic increase with advancing stages of sexual maturity and a decrease
with aging. The age-sensitive testicular circRNAs are highly associated
with spermatogenesis, independent of cognate mRNA expression. The
tissue/age implications of circRNAs suggest that they present unique
physiological functions rather than simply occurring as occasional
by-products of gene transcription”
(Zhou, Xie, Li et al. 2018, doi:10.1261/rna.067132.118).
“Depending on their localization and specific interactions with DNA, RNA,
and proteins, circular RNAs can modulate transcription and splicing,
regulate stability and translation of cytoplasmic mRNAs, interfere with
signaling pathways, and serve as templates for translation in different
biological and pathophysiological contexts”
(Liu and Chen 2022, doi:10.1016/j.cell.2022.04.021).
“Covalently closed, single-stranded circular RNAs can be produced from
viral RNA genomes as well as from the processing of cellular housekeeping
noncoding RNAs and precursor messenger RNAs. Recent transcriptomic
studies have surprisingly uncovered that many protein-coding genes can be
subjected to backsplicing, leading to widespread expression of a specific
type of circular RNAs (circRNAs) in eukaryotic cells ... Some circRNAs
act as noncoding RNAs to impact gene regulation by serving as decoys or
competitors for microRNAs and proteins. Others form extensive networks of
ribonucleoprotein complexes or encode functional peptides that are
translated in response to certain cellular stresses. Overall, circRNAs
have emerged as an important class of RNAmolecules in gene expression
regulation that impact many physiological processes, including early
development, immune responses, neurogenesis, and tumorigenesis”
(Yang, Wilusz and Chen 2022, doi:10.1146/annurev-cellbio-120420-125117).
“Exon–intron circRNAs (EIciRNAs) are a circRNA subclass with retained
introns. Global features of EIciRNAs remain largely unexplored, mainly
owing to the lack of bioinformatic tools. The regulation of intron
retention (IR) in EIciRNAs and the associated functionality also require
further investigation. We developed a framework, FEICP, which efficiently
detected EIciRNAs from high-throughput sequencing (HTS) data. EIciRNAs
are distinct from exonic circRNAs (EcircRNAs) in aspects such as with
larger length, localization in the nucleus, high tissue specificity, and
enrichment mostly in the brain. Deep learning analyses revealed that
compared with regular introns, the retained introns of circRNAs (CIRs)
are shorter in length, have weaker splice site strength, and have higher
GC content. Compared with retained introns in linear RNAs (LIRs), CIRs
are more likely to form secondary structures and show greater sequence
conservation. CIRs are closer to the 5′-end, whereas LIRs are closer to
the 3′-end of transcripts. EIciRNA-generating genes are more actively
transcribed and associated with epigenetic marks of gene activation.
Computational analyses and genome-wide CRISPR screening revealed that
SRSF1 binds to CIRs and inhibits the biogenesis of most EIciRNAs. SRSF1
regulates the biogenesis of EIciLIMK1, which enhances the expression of
LIMK1 in cis to boost neuronal differentiation, exemplifying
EIciRNA physiological function. Overall, our study has developed the
FEICP pipeline to identify EIciRNAs from HTS data, and reveals multiple
features of CIRs and EIciRNAs. SRSF1 has been identified to regulate
EIciRNA biogenesis. EIciRNAs and the regulation of EIciRNA biogenesis
play critical roles in neuronal differentiation”
(Zhong, Yang, Wang et al. 2024, doi:10.1101/gr.278590.123).
- Long thought to result from “errors” in splicing, exonic circular RNAs (ecircRNAs) now look like having significant roles in the organism. A study has shown circular RNAs to be more stable than associated linear mRNAs in vivo, and “in some cases, the abundance of circular molecules exceeded that of associated linear mRNA by >10-fold. By conservative estimate, we identified ecircRNAs from 14.4% of actively transcribed genes in human fibroblasts...These data show that ecircRNAs are abundant, stable, conserved and nonrandom products of RNA splicing that could be involved in control of gene expression” — for example, by acting as competing endogenous RNAs (Jeck, Sorrentino, Wang et al. 2013). See “Competing endogenous RNAs” below.
- “We demonstrate that the [exonic] circular RNA circ-Foxo3 was highly expressed in non-cancer cells and were associated with cell cycle progression. Silencing endogenous circ-Foxo3 promoted cell proliferation. Ectopic expression of circ-Foxo3 repressed cell cycle progression by binding to the cell cycle proteins cyclin-dependent kinase 2 (also known as cell division protein kinase 2 or CDK2) and cyclin-dependent kinase inhibitor 1 (or p21), resulting in the formation of a ternary complex. Normally, CDK2 interacts with cyclin A and cyclin E to facilitate cell cycle entry, while p21 works to inhibit these interactions and arrest cell cycle progression. The formation of this circ-Foxo3-p21-CDK2 ternary complex arrested the function of CDK2 and blocked cell cycle progression” (Du, Yang, Liu et al. 2016, doi:10.1093/nar/gkw027).
- One very long (1500 nucleotides) circular RNA was found to contain 70 binding sites for the miRNA known as miR-7, which has important roles in cancer and Parkinson’s disease. The circular RNA represses miR-7, resulting in increased expression of the targets of miR-7. Changing the balance between these two molecules was shown to alter brain development in zebrafish (Ledford 2013).
- The fact that they can have many binding sites for a given miRNA makes the impact of circular RNAs on gene expression that much greater. And, in the reverse direction: destruction of a circular RNA can release many miRNAs at once, which then can pursue their target mRNAs (Kosik 2013).
- “It is interesting that, whereas the linear competing endogenous RNAs have a short half-life that allows a rapid control of sponge activity, circRNAs have much greater stability and their turnover can be controlled by the presence of a perfectly matched miRNA target site” (Fatica and Bozzoni 2014).
- “Sequence annotation suggests that most circRNAs are generated from splicing in reversed orders across exons ... we constructed a single exon minigene containing split GFP [green flourescent protein], and found that the pre-mRNA indeed produces circRNA through efficient backsplicing in human and Drosophila cells. The backsplicing is enhanced by complementary introns that form double-stranded RNA structure to bring splice sites in proximity, but such structure is not required. Moreover, backsplicing is regulated by general splicing factors and cis-elements, but with regulatory rules distinct from canonical splicing. The resulting circRNA can be translated to generate functional proteins. Unlike linear mRNA, poly-adenosine or poly-thymidine in 3' UTR can inhibit circular mRNA translation. This study revealed that backsplicing can occur efficiently in diverse eukaryotes to generate circular mRNAs” (Wang and Wang 2015, doi:10.1261/rna.048272.114).
- “Strikingly, exon circularization efficiency can be regulated by competition between RNA pairing across flanking introns or within individual introns. Importantly, alternative formation of inverted repeated Alu pairs and the competition between them can lead to alternative circularization, resulting in multiple circular RNA transcripts produced from a single gene”. “Our work shows that alternative circularization coupled with alternative splicing can produce a variety of additional circular RNAs from one gene. Taken together, these lines of evidence imply a new level of complexity in transcriptomes and their regulation” (Zhang, Wang, Zhang et al. 2014, doi:10.1016/j.cell.2014.09.001).
- “We report a class of circRNAs associated with RNA polymerase II in human cells. In these circRNAs, exons are circularized with introns ‘retained’ between exons; we term them exon-intron circRNAs or EIciRNAs. EIciRNAs predominantly localize in the nucleus, interact with U1 snRNP [a ribonucleoprotein involved in RNA splicing] and promote transcription of their parental genes” (Li, Huang, Bao et al. 2015, doi:10.1038/nsmb.2959). The abundance of many circRNAs is fairly low, but the authors point out that where the circRNA acts at the genomic locus from which it is generated, the quantities need not be high in order to be effective.
- “We show that hundreds of circRNAs are regulated during human epithelial-mesenchymal transition (EMT) [a cellular differentiation process important in embryo development] and find that the production of over one-third of abundant circRNAs is dynamically regulated by the alternative splicing factor, Quaking (QKI), which itself is regulated during EMT. Furthermore, by modulating QKI levels, we show the effect on circRNA abundance is dependent on intronic QKI binding motifs. Critically, the addition of QKI motifs is sufficient to induce de novo circRNA formation from transcripts that are normally linearly spliced. These findings demonstrate circRNAs are both purposefully synthesized and regulated by cell-type specific mechanisms, suggesting they play specific biological roles in EMT” (Conn, Pillman, Toubia et al. 2015, doi:10.1016/j.cell.2015.02.014).
- “Production of a single circRNA from the pre-mRNA of the Muscleblind splicing factor was recently shown to be regulated by Muscleblind itself” (Conn, Pillman, Toubia et al. 2015, doi:10.1016/j.cell.2015.02.014, citing work by Ashwal-Fluss et al. 2014).
- “We report the discovery of a class of abundant circular noncoding RNAs that are produced during metazoan tRNA splicing. These transcripts, termed tRNA intronic circular (tric)RNAs, are conserved features of animal transcriptomes. Biogenesis of tricRNAs requires anciently conserved tRNA sequence motifs and processing enzymes, and their expression is regulated in an age-dependent and tissue-specific manner” ( Lu, Filonov, Noto et al. 2015, doi:10.1261/rna.052944.115).
- Circular RNAs and brain development. Circular RNAs are “enriched in the nervous system of both mammals and invertebrates. The reasons for this enrichment seem to be twofold, as circRNAs are derived mainly from linear mRNAs expressed in the nervous system and genes with wider expression patterns are more likely to present a circular variant in the brain. For some of these genes, the circular variant is even the predominant isoform in brain” (Aprea and Calegari 2015, doi:10,.15252/embj.201592655).
- “Brain-expressed circRNAs are differentially expressed among different regions and during mouse development [they show] an overall upregulation during neuronal differentiation. Surprisingly, they are preferentially derived from coding and 5' UTR exons, in particular from host genes involved in synaptic function. Moreover, circRNAs appear enriched in synaptic compartments and show a clear upregulation during development at the onset of synaptogenesis. Thus, circRNAs appear to be particularly relevant for synaptogenesis and synaptic function” (Aprea and Calegari 2015, doi:10,.15252/embj.201592655).
- “Piwecka et al. used CRISPR-Cas9 technology to remove the locus encoding the circular RNA Cdr1as from the mouse genome. Single-cell electrophysiological measurements in excitatory neurons revealed an increase in spontaneous vesicle release from the knockout mice and depression in the synaptic response with two consecutive stimuli, indicating that Cdr1as deficiency leads to dysfunction of excitatory synaptic transmission. Small RNA sequencing of several major regions of the brain showed that expression of two microRNAs, miR-7 and miR-671, that bind to Cdr1as decreased and increased, respectively. These results, along with expression analyses, suggest that neuronal Cdr1as stabilizes or transports miR-7, which in turn represses genes that are early responders to different stimuli” (Piwecka, Glažar, Hernandez-Miranda et al. 2017, doi:10.1126/science.aam8526).
- “We show that tumors harboring chromosomal translocations also harbor circRNAs derived from the rearranged genome: aberrant fusion-circRNAs (f-circRNA). We further show that such f-circRNAs can be functionally relevant and tumor promoting, with potential diagnostic and therapeutic implications” (Guarnerio, Bezzi, Jeong et al. 2016, doi:10.1016/j.cell.2016.03.020).
- “Circular RNAs are generated at low levels from many protein-coding genes. Liu et al. now reveal that many of these transcripts bind and inhibit the double-stranded RNA (dsRNA)-dependent kinase PKR. Upon viral infection, circular RNAs are globally degraded to release PKR, which becomes activated to aid in the immune response” (Wilusz 2019, doi:10.1016/j.cell.2019.04.020).
- “Circular RNAs (circRNAs) have emerged as key regulators of a wide variety of biological processes, but the roles of mitochondrial circRNAs are largely unknown ... Zhao et al. (2020) reveal that mitochondrial DNA-encoded circRNAs interact with ATP synthase subunit β (ATP5B) to inhibit the output of mitochondrial reactive oxygen species and the activation of liver fibroblasts, which regulate the pathogenesis of liver disease” (Yan and Chen 2020, doi:10.1016/j.cell.2020.09.028).
-
Exons
“The processing of RNA transcripts from mammalian genes occurs in
proximity to their transcription. Here, we describe a phenomenon
affecting thousands of genes that we call exon-mediated activation of
transcription starts, in which the splicing of internal exons impacts
promoter choice and the expression level of the gene. We observed that
evolutionary gain of internal exons is associated with gain of new
transcription start sites nearby and increased gene expression.
Inhibiting exon splicing reduced transcription from nearby promoters, and
creation of new spliced exons activated transcription from cryptic
promoters. The strongest effects occurred for weak promoters located
proximal and upstream of efficiently spliced exons. Together, our
findings support a model in which splicing recruits transcription
machinery locally to influence TSS choice and identify exon gain, loss,
and regulatory change as major contributors to the evolution of
alternative promoters and gene expression in mammals”
(Fiszbein, Krick, Begg et al. 2019, doi:10.1016/j.cell.2019.11.002).
-
Introns
Introns are the non-protein-coding portions of a gene normally spliced
out, along with any (protein-coding) exons removed by alternative
splicing. Introns remaining in an mRNA after splicing have long been
thought to be nothing but mistakes. But, as so often happens, such
“mistakes” turn out to have regulatory potential. Likewise, the
excision of an intron can have more significance than just the removal
of “junk”.
- A research group studying normal white blood cell differentiation found intron retention (IR) to be “a physiological mechanism of gene expression control. IR regulates the expression of 86 functionally related genes, including those that determine the nuclear shape that is unique to granulocytes. [Granulocytes are a type of white blood cell.] Retention of introns in specific genes is associated with downregulation of splicing factors and higher GC content. IR, conserved between human and mouse, led to reduced mRNA and protein levels by triggering the nonsense-mediated decay (NMD) pathway. In contrast to the prevalent view that NMD is limited to mRNAs encoding aberrant proteins, our data establish that IR coupled with NMD is a conserved mechanism in normal granulopoiesis. Physiological IR may provide an energetically favorable level of dynamic gene expression control prior to sustained gene translation” (Wong, Ritchie, Ebner et al. 2013).
- “Differentiating erythroblasts execute a dynamic alternative splicing program shown here to include extensive and diverse intron retention (IR) events. Cluster analysis revealed hundreds of developmentally-dynamic introns that exhibit increased IR in mature erythroblasts, and are enriched in functions related to RNA processing such as SF3B1 spliceosomal factor. Distinct, developmentally-stable IR clusters are enriched in metal-ion binding functions and include mitoferrin genes SLC25A37 and SLC25A28 that are critical for iron homeostasis. Some IR transcripts are abundant, e.g. comprising ∼50% of highly-expressed SLC25A37 and SF3B1 transcripts in late erythroblasts, and thereby limiting functional mRNA levels. IR transcripts tested were predominantly nuclear-localized. Splice site strength correlated with IR among stable but not dynamic intron clusters, indicating distinct regulation of dynamically-increased IR in late erythroblasts. Retained introns were preferentially associated with alternative exons with premature termination codons. High IR was observed in disease-causing genes including SF3B1 and the RNA binding protein FUS. Comparative studies demonstrated that the intron retention program in erythroblasts shares features with other tissues but ultimately is unique to erythropoiesis. We conclude that IR is a multi-dimensional set of processes that post-transcriptionally regulate diverse gene groups during normal erythropoiesis, misregulation of which could be responsible for human disease” (Pimentel, Parra, Gee et al. 2016, doi:10.1093/nar/gkv1168).
- “We observed that intron retention [IR] is prevalent in polyadenylated transcripts in resting CD4+ T cells and is significantly reduced upon T cell activation. Several lines of evidence suggest that intron-retained transcripts are less stable than fully spliced transcripts ... Further, the majority of the genes upregulated in activated T cells are accompanied by a significant reduction in IR. Of these 1583 genes, 185 genes are predominantly regulated at the IR level, and highly enriched in the proteasome pathway, which is essential for proper T cell proliferation and cytokine release. These observations were corroborated in both human and mouse CD4+ T cells. Our study revealed a novel post-transcriptional regulatory mechanism that may potentially contribute to coordinated and/or quick cellular responses to extracellular stimuli such as an acute infection” (Ni, Yang, Han et al. 2016, doi:10.1093/nar/gkw591).
- In yeast there is little alternative splicing, and genes that do have introns only have one or two of them. Yet it’s been found that deletion of an intron from one copy of a paralogous (“originating from duplication”) pair of ribosomal protein genes (of which there are many) often regulates the expression not only of the altered gene, but also of the paralog. Expression of the unaltered gene may be either increased or decreased — and not merely in a fashion compensatory to change in the altered gene’s expression. “Introns appear to mediate a variety of regulatory pathways designed to modulate intergenic regulation” (Parenteau, Durand and Morin 2011). It may be, for example, that an mRNA whose intron is not removed by splicing may interact by base pairing with the intron of the paralog gene, and thereby perform a regulatory role.
-
Detained introns. Many specific introns “are significantly
more abundant than the other introns within polyadenylated
transcripts; we classified these as ‘detained’ introns (DIs). We
identified thousands of DIs, many of which are evolutionarily
conserved, in human and mouse cell lines as well as the adult mouse
liver. DIs can have half-lives of over an hour yet remain in the
nucleus and are not subject to nonsense-mediated decay. Drug
inhibition of Clk, a stress-responsive kinase, triggered rapid
splicing changes for a specific subset of DIs; half showed
increased splicing, and half showed increased intron detention,
altering transcript pools of >300 genes ... The splicing of some
DIs ... was also altered following DNA damage ... These data
suggest a widespread mechanism by which the rate of splicing of DIs
contributes to the level of gene expression”.
In sum: “As direct evidence that DIs can contribute to gene regulation, we showed that inhibition of Clk kinase activity as well as DNA damage can modulate the rate of splicing for particular subsets of DIs, enabling coordinated control of specific genes” (Boutz, Bhutkar and Sharp 2015, doi:10.1101/gad.247361.114).
- “Stable intronic sequence RNAs (sisRNAs) are conserved in various organisms. Recent observations in Drosophila suggest that sisRNAs often engage in regulatory feedback loops to control the expression of their parental genes. The use of sisRNAs as mediators for local feedback control may be a general phenomenon.” (Pek 2018, doi:10.1016/j.tig.2018.01.006)
-
RNA editing
RNA editing occurs when individual bases of a pre-mRNA (precursor
messenger RNA) are altered by editing enzymes.
- “Here,we report a new mechanism for the functionality of RNA editing — a crosstalk with PIWI-interacting RNA (piRNA) biogenesis. [In the rhesus macaque] we deciphered accurate RNA editome across both long transcripts and the piRNA species. Superimposing and comparing these two distinct RNA editome profiles revealed 4,170 editing-bearing piRNA variants, or epiRNAs, that primarily derived from edited long transcripts. These epiRNAs represent distinct entities that evidence an intersection between RNA editing regulations and piRNA biogenesis ... these findings are consistent in human, supporting the conservation of this mechanism during the primate evolution. Overall, our study reports the earliest lines of evidence for a crosstalk between selectively constrained RNA editing regulation and piRNA biogenesis, and further illustrates that such an interaction may contribute substantially to the diversification of the piRNA repertoire in primates.” (Yang, Chen, Liu et al. 2015, doi:10.1093/molbev/msv183)
- RNA editing and RNA splicing: “By sequencing the RNA of different subcellular fractions, we examined the timing of adenosine-to-inosine (A-to-I) RNA editing and its impact on alternative splicing. We observed that >95% A-to-I RNA editing events occurred in the chromatin-associated RNA prior to polyadenylation. We report about 500 editing sites in the 3′ acceptor sequences that can alter splicing of the associated exons. These exons are highly conserved during evolution and reside in genes with important cellular function. Furthermore, we identified a second class of exons whose splicing is likely modulated by RNA secondary structures that are recognized by the RNA editing machinery. The genome-wide analyses, supported by experimental validations, revealed remarkable interplay between RNA editing and splicing and expanded the repertoire of functional RNA editing sites” (Hsiao, Bahn, Yang et al. 2018, doi:10.1101/gr.231209.117).
-
A-to-I editing
The most common form of editing results in adenosine being
changed to inosine (A-to-I editing), which in turn is
interpreted as guanosine during translation. This can change
the protein encoded by the mRNA, and so serves to diversify the
proteins in an organism. Editing occurs in stretches of an RNA
that are folded into a duplex — that is, where base-pairing has
taken place. “A-to-I editing alters RNA structure, coding
potential, splicing pattern, or cellular distribution, and
offers a means to regulate gene expression at a variety of
post-transcriptional levels” (Mao, Zhang and Spector 2011).
There are hundreds of sites liable to editing in RNAs (Lindberg
and Lundeberg 2009), and editing can occur in
noncoding as well as coding regions of an RNA. “Bioinformatic
analyses predicted that >5% of human mRNAs contain editing
sites in noncoding sequences”. The presence of an editing
enzyme (e.g., ADAR1) prevents cell death and is essential for
organism survival (Vitali and Scadden 2010).
For a given RNA subject to editing, “the fraction of edited
molecules ranges from a few to almost 100% of [a] gene’s
transcripts. Thus, edited and unedited variants are usually
coexpressed within the same cell providing for transcriptome
variation without the all-or-nothing effect of DNA mutations in
the genome” (Farajollahi and Maas 2010).
“We systematically characterized the miRNA editing profiles of 8595
samples across 20 cancer types ... and identified 19
adenosine-to-inosine (A-to-I) RNA editing hotspots. These miRNA
editing events show extensive correlations with key clinical variables
(e.g., tumor subtype, disease stage, and patient survival time) and
other molecular drivers. Focusing on the RNA editing hotspot in
miR-200b, a key tumor metastasis suppressor, we found that the
miR-200b editing level correlates with patient prognosis opposite to
the pattern observed for the wild-type miR-200b expression. We further
experimentally showed that, in contrast to wild-type miRNA, the edited
miR-200b can promote cell invasion and migration through its impaired
ability to inhibit ZEB1/ZEB2 and acquired concomitant ability to
repress new targets, including LIFR, a well-characterized metastasis
suppressor. Our study highlights the importance of miRNA editing in
gene regulation and suggests its potential as a biomarker for cancer
prognosis and therapy”
(Wang, Xu, Yu et al. 2017, doi:10.1101/gr.219741.116).
“Modifications of RNA affect its function and stability. RNA editing
is unique among these modifications because it not only alters the
cellular fate of RNA molecules but also alters their sequence relative
to the genome. The most common type of RNA editing is A-to-I editing
by double-stranded RNA-specific adenosine deaminase (ADAR) enzymes.
Recent transcriptomic studies have identified a number of ‘recoding’
sites at which A-to-I editing results in non-synonymous substitutions
in protein-coding sequences. Many of these recoding sites are
conserved within (but not usually across) lineages, are under positive
selection and have functional and evolutionary importance. However,
systematic mapping of the editome across the animal kingdom has
revealed that most A-to-I editing sites are located within mobile
elements in non-coding parts of the genome. Editing of these
non-coding sites is thought to have a critical role in protecting
against activation of innate immunity by self-transcripts”
(Eisenberg and Levanon 2018, doi:10.1038/s41576-018-0006-1).
Reseach indicates “that editing is regulated in a [RNA] site-specific
manner by the different interplay between ADAR1 and ADAR2”
(Cruz, Yuki Kato, Taisuke Nakahama 2020, doi:10.1261/rna.072728.119).
“We uncover a molecular mechanism that regulates RNA editing in a
neural- and development-specific manner. Comparing editomes during
development led to the identification of neural transcripts that were
edited only in one life stage. The stage-specific editing is largely
regulated by differential gene expression during neural development.
Proper expression of nearly one-third of the neurodevelopmentally
regulated genes is dependent on adr-2, the sole A-to-I editing enzyme
in C. elegans. However, we also identified a subset of neural
transcripts that are edited and expressed throughout development.
Despite a neural-specific down-regulation of adr-2 during development,
the majority of these sites show increased editing in adult neural
cells. Biochemical data suggest that ADR-1, a deaminase-deficient
member of the adenosine deaminase acting on RNA (ADAR) family, is
competing with ADR-2 for binding to specific transcripts early in
development. Our data suggest a model in which during neural
development, ADR-2 levels overcome ADR-1 repression, resulting in
increased ADR-2 binding and editing of specific transcripts. Together,
our findings reveal tissue- and development-specific regulation of RNA
editing and identify a molecular mechanism that regulates ADAR
substrate recognition and editing efficiency”
(Rajendren, Dhakal, Vadlamani et al. 2021, doi:10.1101/gr.267575.120).
“Modified bases act as marks on cellular RNAs so that they can be
distinguished from foreign RNAs, reducing innate immune responses to
endogenous RNA. In humans, mutations giving reduced levels of one base
modification, adenosine-to-inosine deamination, cause a viral
infection mimic syndrome, a congenital encephalitis with aberrant
interferon induction”
(Quin, Sedmík, Vukić et al. 2021, doi:10.1016/j.tibs.2021.02.002).
- It has now been found that the tertiary structure of an RNA can be decisive for A-to-I editing. That is, a small bulge in the three-dimensional shape of the folded RNA — perhaps resulting from a single-letter change — can be decisive for the editing process. “A single synonymous substitution might result in a nearly complete loss of editing” (Tian, Yang, Sachsenmaier et al. 2011) — or an opposite substitution could result in a gain of editing. In other words, a one-letter “synonymous” change in the DNA code — a change that supposedly doesn’t specify any difference in the protein coded for — can alter the tertiary structure of the associated RNA such that, through editing or its loss, the RNA now produces a different protein. Of course, all the other factors affecting RNA folding may also play a above. (See “RNA folding” above and RNA structure below.)
- “Drosophila and rodents use editing to fine tune protein function temporally, over the course of development and spatially, in different brain regions” (Garrett and Rosenthal 2012).
- “Our results show that A-to-I RNA editing is widespread in the brain transcriptomes of humans and non-human primates”. “The most intriguing finding of our study is a general increase in RNA-editing levels in the brains of humans and non-human primates with advanced age. These results match the editing level increase with age reported in the mouse brain”. “The mis-regulation of A-to-I RNA editing has been shown to affect neural functions in various organisms from humans to worms. A-to-I editing affects not only the protein sequences themselves, but also RNA stability, cellular localization, splicing, and translation efficiency”. “Overall, substantial conservation of RNA-editing patterns among species and brain regions, the presence of a common trend for RNA-editing increase with advanced age, as well as greater sequence conservation of sites showing age-related increase at the genome sequence level indicate that RNA editing may play substantial functional roles in the primate and mammalian brains” (Li, Bammann, Li et al. 2013).
- “A growing body of evidence has linked RNA editing to the small ncRNA species of miRNAs, alterations of which are known to have developmental and pathological implications”. (Yang, Chen, Liu et al. 2015, doi:10.1093/molbev/msv183)
- miRNAs can also undergo A-to-I editing. This can include editing of their seed sequences, which determine what mRNAs will be targeted by the miRNAs. In mice: “We show that increased editing during development gradually changes the proportions of the two miR-376a isoforms, which previously have been shown to have different targets. Several other miRNAs that also are edited in the seed sequence show an increased level of editing through development. By comparing editing of pri-miRNA with editing and expression of the corresponding mature miRNA, we also show an editing-induced developmental regulation of miRNA expression. Taken together, our results imply that RNA editing influences the miRNA repertoire during brain maturation” (Ekdahl, Farahani, Behm et al. 2012).
- “Our analysis identified some U2/U12-like non-canonical splice sites that are converted into canonical splice sites by RNA A-to-I editing” (Torella, Li, Kinrade et al. 2014, doi:10.1093/nar/gku744).
- In a study of Caenorhabditis elegans, which has two ADARs, ADR-1 and ADR-2: “A total of 99.5% of the 47,660 A-to-I editing sites were found in clusters. Of the 3080 editing clusters, 65.7% overlapped with DNA transposons in noncoding regions and 73.7% could form hairpin structures. The numbers of editing sites and clusters were highest at the L1 and embryonic stages. The editing frequency of a cluster positively correlated with the number of editing sites within it. Intriguingly, for 80% of the clusters with 10 or more editing sites, almost all expressed transcripts were edited. Deletion of adr-1 reduced the editing frequency but not the number of editing clusters, whereas deletion of adr-2 nearly abolished RNA editing, indicating a modulating role of ADR-1 and an essential role of ADR-2 in A-to-I editing. Quantitative proteomics analysis showed that adr-2 mutant worms altered the abundance of proteins involved in aging and lifespan regulation. Consistent with this finding, we observed that worms lacking RNA editing were short-lived. Taken together, our results reveal a sophisticated landscape of RNA editing and distinct modes of action of different ADARs” (Zhao, Zhang, Gao et al. 2015, doi:10.1101/gr.176107.114).
- “In primates, IRAlus [inverted repeat Alus] are the main binding site for ADARs and are subject to editing at multiple sites. More than 90% of A-to-I editing in humans occurs within Alu elements. Multisite A-to-I editing within exonized Alu elements are predicted to result in amino acid recoding, because inosines are recognized as guanosines by translating ribosomes. A-to-I editing within intronic IRAlus can generate new splice sites that lead to the exonization of Alu elements (Elbarbary, Lucas and Maquat 2016, doi:10.1126/science.aac7247).
- ADAR1-null mice die in utero owing to failed erythropoiesis and liver disintegration. A research group confirmed that this is due to defects in RNA editing. “The absence of RNA editing led to upregulation of interferon-stimulated genes, similar to those activated in vitro by dsRNAs containing adenosine, but not inosine, demonstrating that editing by ADAR1 suppresses the interferon response in homeostatic conditions. Many editing sites were found in the 3' UTRs of three erythropoiesis genes; these were predicted to form long dsRNA stretches in unedited but not in edited transcripts. Knocking out MDA5 — which is a sensor of viral dsRNA and activator of the interferon response — in [the ADAR1-null] mice rescued their phenotype. Thus, sensing of unedited endogenous dsRNAs by MDA5 activates erythropoiesis-detrimental interferon responses” (Zlotorynski 2015, doi:10.1038/nrm4050).
- “Adenosine-to-inosine RNA editing by ADARs affects thousands of adenosines in an organism's transcriptome. However, adenosines are not edited at equal levels nor do these editing levels correlate well with ADAR expression levels. Therefore, additional mechanisms are utilized by the cell to dictate the editing efficiency at a given adenosine ... We demonstrate [in Caenorhabditis elegans] that a double-stranded RNA (dsRNA) binding protein, ADR-1, inhibits editing in neurons ... Furthermore, expression of ADR-1 and mRNA expression of the editing target can act synergistically to regulate editing efficiency. In addition, we identify a dsRNA region within the Y75B8A.8 3' UTR that acts as a cis-regulatory element by enhancing ADR-2 editing efficiency” (Washburn and Hundley 2016, doi:10.1261/rna.055079.115).
- The following is not a matter of RNA editing, but illustrates a ubiquitous truth of molecular biology: molecules central to one particular function are found also playing roles in altogether unrelated (or seemingly unrelated) functions: “By using different cell-culture based retrotransposition assays in HeLa cells, we demonstrated a novel function of ADAR1 as suppressor of L1 retrotransposition. Apparently, this inhibitory mechanism does not occur through ADAR1 editing activity. Furthermore, we showed that ADAR1 binds the basal L1 RNP complex. Overall, these data support the role of ADAR1 as regulator of L1 life cycle” (Orecchini, Doria, Antonioni 2016, doi:10.1093/nar/gkw834).
- “We identify a regulatory mechanism whereby ADAR2 enhances target RNA stability by limiting the interaction of RNA-destabilizing proteins with their cognate substrates” (Anantharaman, Tripathi, Abid Khan et al. 2017, doi:10.1093/nar/gkw1304).
- “Both p150 and p110 isoforms of ADAR1 convert adenosine to inosine in double-stranded RNA (dsRNA). ADAR1p150 suppresses the dsRNA-sensing mechanism that activates MDA5–MAVS–IFN signaling in the cytoplasm ... Here, we show that stress-activated phosphorylation of ADAR1p110 by MKK6-p38-MSK MAP kinases promotes its binding to Exportin-5 and its export from the nucleus. After translocating to the cytoplasm, ADAR1p110 suppresses apoptosis in stressed cells by protecting many antiapoptotic gene transcripts that contain 3'-untranslated-region dsRNA structures primarily comprising inverted Alu repeats. ADAR1p110 competitively inhibits binding of Staufen1 to the 3'-untranslated-region [of] dsRNAs and antagonizes Staufen1-mediated mRNA decay” (Sakurai, Shiromoto, Ota et al. 2017, doi:10.1038/nsmb.3403).
- “in Caenorhabditis elegans, A-to-I editing in double-stranded regions of protein-coding transcripts protects these RNAs from targeting by the RNAi pathway. Disruption of this safeguard through loss of ADAR activity coupled with enhanced RNAi results in developmental abnormalities and profound changes in gene expression that suggest aberrant induction of an antiviral response. Thus, editing of cellular dsRNA by ADAR helps prevent host RNA silencing and inadvertent antiviral activity” (Pasquinelli 2018, doi:10.1101/gad.313049.118).
- “Recognition of dsRNA [double-stranded RNA] molecules activates the MDA5-MAVS pathway, and plays a critical role in stimulating type-I interferon responses in psoriasis. However, the source of the dsRNA accumulation in psoriatic keratinocytes remains largely unknown. A-to-I RNA editing is a common co- or post-transcriptional modification that diversifies adenosine in dsRNA, and leads to unwinding of dsRNA structures. Thus, impaired RNA editing activity can result in an increased load of endogenous dsRNAs. Here we provide a transcriptome wide analysis of RNA editing across dozens of psoriasis patients, and demonstrate a global editing reduction in psoriatic lesions. In addition to the global alteration, we also detect editing changes in functional recoding sites located in the IGFBP7, COPA, and FLNA genes. Accretion of dsRNA activates autoimmune responses therefore the results presented here, linking for the first time an autoimmune disease to reduction in global editing level, are relevant to a wide range of autoimmune diseases. (Shallev, Kopel, Feiglin et al. 2018, doi:10.1261/rna.064659.117).
- Antarctic and tropical octopuses that produce proteins from almost identical rectifier K+ genes were found to modify the gene transcripts extensively through A-to-I RNA editing in order to express proteins adapted to the temperature differences of their environments. Thus, “RNA editing can respond to an external pressure: temperature” (Garrett and Rosenthal 2012).
- “In ectothermic organisms, including Drosophila and Cephalopoda, where body temperature mirrors ambient temperature, decreases in environmental temperature lead to increases in A-to-I RNA editing and cause amino acid recoding events that are thought to be adaptive responses to temperature fluctuations. In contrast, endothermic mammals, including humans and mice, typically maintain a constant body temperature despite environmental changes. Here, A-to-I editing primarily targets repeat elements, rarely results in the recoding of amino acids, and plays a critical role in innate immune tolerance. Hibernating ground squirrels provide a unique opportunity to examine RNA editing in a heterothermic mammal whose body temperature varies over 30°C and can be maintained at 5°C for many days during torpor. We profiled the transcriptome in three brain regions at six physiological states to quantify RNA editing and determine whether cold-induced RNA editing modifies the transcriptome as a potential mechanism for neuroprotection at low temperature during hibernation. We identified 5165 A-to-I editing sites in 1205 genes with dynamically increased editing after prolonged cold exposure. The majority (99.6%) of the cold-increased editing sites are outside of previously annotated coding regions, 82.7% lie in SINE-derived repeats, and 12 sites are predicted to recode amino acids. Additionally, A-to-I editing frequencies increase with increasing cold-exposure, demonstrating that ADAR remains active during torpor. Our findings suggest that dynamic A-to-I editing at low body temperature may provide a neuroprotective mechanism to limit aberrant dsRNA accumulation during torpor in the mammalian hibernator” (Riemondy, Gillen, White et al. 2018, doi:10.1261/rna.066522.118).
-
APOBEC1 (C-to-U) editing
“C-to-U DNA editing enables [the proteins that do the editing] to
inhibit parasitic viruses and retrotransposons by disrupting their
genomic content. In addition to attacking genomic invaders, APOBECs
can target their host genome, which can be beneficial by initiating
processes that create antibody diversity needed for the immune system
or by accelerating the rate of evolution. AID can also alter gene
regulation by removing epigenetic modifications from genomic DNA.
However, when uncontrolled, these powerful agents of change can
threaten genome stability and eventually lead to cancer”
(Knisbacher, Gerber and Levanon 2016, doi:10.1016/j.tig.2015.10.005).
“The AID/APOBEC polynucleotide cytidine deaminases have historically
been classified as either DNA mutators or RNA editors based on their
first identified nucleic acid substrate preference. DNA mutators can
generate functional diversity at antibody genes but also cause genomic
instability in cancer. RNA editors can generate informational
diversity in the transcriptome of innate immune cells, and of cancer
cells. Members of both classes can act as antiviral restriction
factors. Recent structural work has illuminated differences and
similarities between AID/APOBEC enzymes that can catalyse DNA
mutation, RNA editing or both, suggesting that the strict functional
classification of members of this family should be reconsidered”
(Pecori, Di Giorgia, Lorenzo, and Papavasiliou 2022,
doi:10.1038/s41576-022-00459-8).
- At first known to affect only a single mRNA in mammals, C-to-U editing by the APOBEC1 protein has now been described in 32 additional cases. It affects the 3'-UTR region of the mRNAs, typically in highly conserved segments, where it could influence regulation by miRNAs, polyadenylation, subcellular localization of the mRNA, and translational efficiency, as well as the binding of various regulatory proteins (Rosenberg, Hamilton, Mwangi et al. 2012).
-
Pseudo-uridylation
Pseudo-uridylation is the change of a uridine in an RNA to
pseudouridine (a rotation isomer of uridine). “When incorporated
into RNA, pseudouridine can alter RNA structure, increase base
stacking, improve base-pairing, and rigidify the sugar-phosphate
backbone. Studies have also linked pseudouridine, either directly
or indirectly, to human disease. ... Owing to its unique structural
and chemical properties and its proven biological relevance,
pesudouridine has increasingly attracted research attention” (Ge
and Yu 2013).
Recent findings reveal “that pseudouridylation is a dynamic and
regulated process that is induced in response to cell state”. “The
function of pseudouridylation is best understood within the context of
mRNA splicing and translation, as both the spliceosomal small nuclear
RNAs (snRNAs; key components of the spliceosome) and the ribosomal
RNAs (key components of the ribosome) are abundantly pseudouridylated.
In fact, pseudouridine residues are concentrated in evolutionarily
conserved and functionally important regions of these RNAs, with
implications for the primary, secondary and tertiary structures of the
molecules. Indeed, experimental data have established the importance
of pseudouridylation in rRNA and spliceosomal small nuclear
ribonucleoprotein (snRNP) biogenesis, efficiency of pre-mRNA splicing
and translation fidelity” (Karijolich, Yi and Yu 2015,
doi:10.1038/nrm4040).
“Recent advances in pseudouridine detection reveal a complex
pseudouridine landscape that includes messenger RNA and diverse
classes of noncoding RNA in human cells. The known molecular functions
of pseudouridine, which include stabilizing RNA conformations and
destabilizing interactions with varied RNA-binding proteins, suggest
that RNA pseudouridylation could have widespread effects on RNA
metabolism and gene expression. Here, we emphasize how much remains to
be learned about the RNA targets of human pseudouridine synthases,
their basis for recognizing distinct RNA sequences, and the mechanisms
responsible for regulated RNA pseudouridylation. We also examine the
roles of noncoding RNA pseudouridylation in splicing and translation
and point out the potential effects of mRNA pseudouridylation on
protein production, including in the context of therapeutic mRNAs”
(Borchardt, Martinez and Gilbert 2020,
doi:10.1146/annurev-genet-112618-043830).
- “Environmental stimuli induce pseudouridylation of spliceosomal small nuclear RNA U2 at novel sites impacting pre-mRNA splicing. After the regulatory modification of protein and of DNA, that of RNA now adds another level of complexity to the cellular signaling landscape” (Meier 2011).
- Pseudo-uridylation is also found in tRNAs and rRNAs. It is not now known whether or to what degree it occurs in protein-coding RNAs (mRNAs), although various studies suggest that “naturally occuring mRNA pseudouridylation is likely to be widespread” (Ge and Yu 2013). Further, it has been shown that if the initial uridine in any of the three mRNA stop codons is changed to a pseudouridine, then, remarkably, the stop codon not only ceases to function as such during translation, but in each case two amino acids are added to the protein being produced. In other words, the genetic code is altered by this modification (Parisien, Yi and Pan 2012). “Given the large number of U-containing sense codons (34 of the 61 sense codons contain one or more uridines), targeted mRNA pseudouridylation portends an expansion of the genetic code” (Ge and Yu 2013).
- “In addition to known pseudouridines in non-coding RNAs, [two new studies] identified hundreds of unknown sites of pseudouridylation in both non-coding and coding RNAs of yeast cells and human cells. These sites were found to be under dynamic regulation, for example, in response to stress such as nutrient deprivation or heat shock ... Promising avenues of investigation are the impact of pseudouridylation on gene regulation, whether through effects on translation, mRNA stability or RNA localization, as well as the mechanisms underlying the dynamic regulation of mRNA pseudouridines in response to stress and developmental cues”. One of the study authors, Wendy Gilbert, remarks that “Perhaps the most exciting prospect is regulated 'rewiring' of the genetic code” (Koch 2014, doi:10.1038/nrg3834).
- “Recently, the first transcriptome-wide maps of RNA pseudouridylation were published, greatly expanding the catalogue of known pseudouridylated RNAs. These data have further implicated RNA pseudouridylation in the cellular stress response and, moreover, have established that mRNAs are also targets of pseudouridine synthases, potentially representing a novel mechanism for expanding the complexity of the cellular proteome” (Karijolich, Yi and Yu 2015, doi:10.1038/nrm4040).
- “Pseudouridylation (Ψ) is the most abundant and widespread type of RNA epigenetic modification in living organisms ... Here, we show that a Ψ-driven posttranscriptional program steers translation control to impact stem cell commitment during early embryogenesis. Mechanistically, the Ψ ‘writer’ PUS7 modifies and activates a novel network of tRNA-derived small fragments (tRFs) targeting the translation initiation complex. PUS7 inactivation in embryonic stem cells impairs tRF-mediated translation regulation, leading to increased protein biosynthesis and defective germ layer specification. Remarkably, dysregulation of this posttranscriptional regulatory circuitry impairs hematopoietic stem cell commitment and is common to aggressive subtypes of human myelodysplastic syndromes” (Guzzi, Cieśla, Ngoc et al. 2018, doi:10.1016/j.cell.2018.03.008).
- “Although their sequences differ, eukaryotic box H/ACA RNAs [a group of small RNAs] all share the same unique hairpin-hinge-hairpin-tail structure. Almost all of them function as guides that primarily direct pseudouridylation of rRNAs and spliceosomal snRNAs at specific sites ... Here, we ... identify the minimum number of base pairs (8), required for RNA-guided pseudouridylation. In addition, we find that the pseudouridylation pocket, present in each hairpin of box H/ACA RNA, exhibits flexibility in fitting slightly different substrate sequences. Our results are consistent across three independent pseudouridylation pockets tested, suggesting that our findings are generally applicable to box H/ACA RNA-guided RNA pseudouridylation” (Zoysa, Wu, Katz and Yu 2018, doi:10.1261/rna.066837.118).
- “Pseudouridine is the most abundant modified nucleotide in RNA, but the role of mRNA pseudouridylation is unclear. Martinez et al. show that pre-mRNA pseudouridylation can broadly modulate alternative splicing, and identify human pre-mRNA-targeting pseudouridylate synthase (PUS) enzymes” (Zlotorynski 2022, doi:10.1038/s41580-022-00458-x)
-
RNA modifications
“After transcription, mRNA function is largely determined by interactions
between the transcript and trans-acting factors, such as RNA-binding
proteins (RBPs). These interactions can be further regulated by RNA
post-transcriptional modifications. The presence or absence of these
modifications can act as a scaffold where specialized RBPs bind,
depending on their affinity for the modification. By adjusting the extent
to which an RNA is modified, cellular signaling pathways can leverage RNA
modifications to influence gene expression without changing genomic
information” (Chan and Batista 2020, doi:10.1038/s41588-020-0685-3).
“Well over 100 different modifications decorate nucleotides in cellular
RNA”. These modifications (unlike, say, methylation of DNA) have long
been more or less ignored, because they were taken to be “constitutive”
(always present in the same way) and therefore irrelevant to gene
regulation. Now this is changing (Meier 2011; Motorin, Lyko and Helm
2010). “Modified nucleosides play an important role in RNA function
and have been identified in multiple RNA types, including tRNAs, rRNAs,
mRNAs and small regulatory RNAs” (Squires and Preiss 2010). Also in
long noncoding RNAs.
“Many of these post-transcriptional modifications are reversible and,
given the range of modifications and targets, may comprise an
additional layer of post-transcriptional regulation analogous to the
epigenetic landscape” (Mercer and Mattick 2013).
“The deposition of chemical modifications into RNA is a crucial regulator
of temporal and spatial gene expression programs during development.
Accordingly, altered RNA modification patterns are widely linked to
developmental diseases. Recently, the dysregulation of RNA modification
pathways also emerged as a contributor to cancer. By modulating cell
survival, differentiation, migration and drug resistance, RNA
modifications add another regulatory layer of complexity to most aspects
of tumourigenesis”
(Delaunay and Frye 2019, doi:10.1038/s41556-019-0319-0).
“RNA is not an exact copy of DNA: processing steps such as splicing,
editing, and base and sugar modification distinguish RNA sequences from
their DNA templates. These modifications, including 140 known modified
ribonucleotides and counting, influence RNA structure and function by
affecting how the RNA interacts with other nucleic acids and regulatory
proteins. Yet, how all the modified nucleotides are distributed in RNA
transcripts remains unknown. This information gap stems from the lack of
methods to sequence full-length RNAs directly. The technology that we
call RNA sequencing is misleading; instead, a more accurate term would be
complementary DNA (cDNA) sequencing, because RNAs are converted back to
DNA by reverse transcription and then sequenced. In the RNA-to-DNA
conversion, important information on nucleotide modifications is lost. No
existing technology can determine the identity and position of all
modifications simultaneously in full-length RNAs at both the
single-molecule and transcriptome-wide scales. The reliance on cDNA
sequences has led to a failure to obtain and understand key regulatory
codes in the RNome of human cells and other organisms, including many
infectious viruses with RNA genomes ... Base and sugar modifications, as
well as splicing, affect RNA chemical properties, topology and function.
Knowledge of the types and locations of the modifications and splice
sites, their extent and their interrelationships is necessary for a basic
understanding of how nucleic acids regulate cellular and organismal
function and how dysregulation leads to diseases.
“Defects in RNA modifications (which are distinct from splice-site alterations) account for more than 100 human diseases, including childhood-onset multiorgan failures, cancers and neurologic disorders. These conditions are now referred to as ‘RNA modopathies’. This number is likely to represent only a small percentage of the actual number of existing RNA modopathies” (Alfonso, Brown, Byers et al. 2021, doi:10.1038/s41588-021-00903-1).
“Studies of the role that [mRNA] modifications play in translation paint
a complex picture. When examining direct regulation of translation, it
appears that the same modification can either promote or repress
translation of an mRNA, depending on the location of the modification or
the biological system studied. Indirectly, the same modification can
either increase mRNA stability, as in the case of hypoxia-induced
stabilization of mRNA, or decrease it, as exemplified in the widely
studied YTHDF2-mediated decay of m6A-methylated mRNA. These
contrasting effects ultimately have different consequences for
translation output. With m6A, the most studied modification,
we are now beginning to understand that the delicate balance of
methylation and demethylation is involved in complex biological processes
such as differentiation and the stress response. Alterations to that
balance contribute to different pathologies. The epitranscriptome,
consisting of various RNA modifications, is thus beginning to be
unraveled as a complex layer of information with major implications for
the regulation of translation in healthy and disease states”
(Peer, Moshitch-Moshkovitz, Rechavi and Dominissini 2019,
doi:10.1101/cshperspect.a032623).
“Until recently, the role of N1-methyladenine (m1A) on mRNAs
during acute stress response remains largely unknown. Here we show that
the methyltransferase complex TRMT6/61A, which generates the
m1A tag, is involved in transcriptome protection during heat
shock. Our bioinformatics analysis indicates that occurrence of the
m1A motif is increased in mRNAs known to be enriched in SGs
[stress granules]. Accordingly, the m1A-generating
methyltransferase TRMT6/61A accumulated in SGs and mass spectrometry
confirmed enrichment of m1A in the SG RNAs. The insertion of a
single methylation motif in the untranslated region of a reporter RNA
leads to more efficient recovery of protein synthesis from that
transcript after the return to normal temperature. Our results
demonstrate far-reaching functional consequences of a minimal RNA
modification on N1-adenine during acute proteostasis stress”
(Alriquet, Calloni, Martínez-Limón et al. 2021, doi:10.1093/jmcb/mjaa023).
“Recent studies suggest noncoding RNAs interact with genomic DNA, forming
RNA•DNA-DNA triple helices, as a mechanism to regulate transcription. One
way cells could regulate the formation of these triple helices is through
RNA modifications. With over 140 naturally occurring RNA modifications,
we hypothesize that some modifications stabilize RNA•DNA-DNA triple
helices while others destabilize them. Here, we focus on a
pyrimidine-motif triple helix composed of canonical U•A-T and C•G-C base
triples. We ... examine how 11 different RNA modifications at a single
position in an RNA•DNA-DNA triple helix affect stability: Compared to the
unmodified U•A-T base triple, some modifications have no significant
change in stability, some have ∼2.5-fold decreases in stability,
and some completely disrupt triple helix formation”
(Kunkler, Schiefelbein, O’Leary et al. 2022, doi:10.1261/rna.079244.122).
Summary: “The main achievement in the field [of RNA modifications]
is the uncovering of a new, intricate, highly sensitive, tuneable layer
of gene expression regulation by mRNA modifications. This new layer of
regulation operates by taking advantage of the unique characteristics of
mRNA — namely, that it is short-lived, highly structured, mobile between
cellular compartments and amplified through transcription. These effects
are mediated in part by ‘readers’, which are exemplified by
methyl-specific binding proteins ... Regulation of gene expression is
also tuned by an interplay between the installation and removal of the
modifications by ‘writers’ and ‘erasers’. Several major lessons have
emerged in the past decade. First, mRNA modifications are highly
prevalent with thousands of gene transcripts modified. Interestingly,
some modifications cluster in specific transcript locations; for example,
inosines are found mostly in repetitive Alu sequences, m6A
preferentially decorates the stop codon vicinity and extremely large
internal exons, and m1A clusters around the AUG start codon,
suggesting that each modification acts through a different mode of
action. Moreover, some modifications, such as m6A and
m1A exhibit high conservation between humans and mice.
“Another important achievement is the discovery that a specific modification can act through different modes of action, through various readers, in a context-dependent manner. An additional important finding is the dynamic nature of some mRNA modifications that allows for a quick response to environmental stimuli; this dynamic nature has already been demonstrated for m6A and m1A. The central role of mRNA modifications is reflected by the devastating effects of aberrant modifications on early development both in humans and mice, as well as in human cancer, inflammation and neurodegeneration, further emphasizing the importance of this regulatory layer” (Gideon Rechavi, quoted in doi:10.1038/nrg.2016.47).
“RNA modifications have recently emerged as an important regulatory layer
of gene expression. The most prevalent and reversible modification on
messenger RNA (mRNA), N6-methyladenosine, regulates most steps of RNA
metabolism and its dysregulation has been associated with numerous
diseases. Other modifications such as 5-methylcytosine and
N1-methyladenosine have also been detected on mRNA but their abundance is
lower and still debated. Adenosine to inosine RNA editing is widespread
on coding and non-coding RNA and can alter mRNA decoding as well as
protect against autoimmune diseases. 2′-O-methylation of the ribose and
pseudouridine are widespread on ribosomal and transfer RNA and contribute
to proper RNA folding and stability. While the understanding of the
individual role of RNA modifications has now reached an unprecedented
stage, still little is known about their interplay in the control of gene
expression. In this review we discuss the examples where such interplay
has been observed and speculate that with the progress of mapping
technologies more of those will rapidly accumulate”
(Worpenberg, Paolantoni and Roignant 2022, doi:10.1002/bies.202100174).
- “The two most abundant mRNA modifications — pseudouridine (Ψ) and N6-methyladenosine (m6A) — affect diverse cellular processes including mRNA splicing, localization, translation, and decay and modulate RNA structure. Here, we test the hypothesis that RNA modifications directly affect interactions between RNA-binding proteins and target RNA. We show that Ψ and m6A weaken the binding of the human single-stranded RNA binding protein Pumilio 2 (hPUM2) to its consensus motif, with individual modifications having effects up to approximately threefold and multiple modifications giving larger effects. While there are likely to be some cases where RNA modifications essentially fully ablate protein binding, here we see modest responses that may be more common. Such modest effects could nevertheless profoundly alter the complex landscape of RNA:protein interactions” (Vaidyanathan, AlSadhan, Merriman et al. 2017, doi:10.1261/rna.060053.116).
-
mRNA adenosine methylation (m6A and m1A)
DNA methylation has for some time been recognized as a major player
in gene regulation. Adenosine methylation in RNA is now gaining
the same recognition. One of the authors of the study by Meyer et
al. cited below remarks, “This finding rewrites the fundamental
concepts of the composition of mRNA because, for 50 years, no one
thought mRNA contained internal modifications that control
function” (ScienceDaily 2012).
“Over 100 types of chemical modifications have been identified in
cellular RNAs. While the 5' cap modification and the poly(A) tail of
eukaryotic mRNA play key roles in regulation, internal modifications
are gaining attention for their roles in mRNA metabolism. The most
abundant internal mRNA modification is N6-methyladenosine
(m6A), and identification of proteins that install,
recognize, and remove this and other marks have revealed roles for
mRNA modification in nearly every aspect of the mRNA life cycle, as
well as in various cellular, developmental, and disease processes.
Abundant noncoding RNAs such as tRNAs, rRNAs, and spliceosomal RNAs
are also heavily modified and depend on the modifications for their
biogenesis and function. Our understanding of the biological
contributions of these different chemical modifications is beginning
to take shape, but it’s clear that in both coding and noncoding RNAs,
dynamic modifications represent a new layer of control of genetic
information”
(Roundtree, Evans, Pan and He 2017, doi:10.1016/j.cell.2017.05.045).
[Referring to work by Fustin, Doi, Yamaguchi et al. 2013:] “Perhaps
the more significant outcome of this study is to demonstrate a
cellular function of m6A methylation: namely to regulate
the nuclear processing of mRNA. This means that RNA can be modified
to carry more information beyond its familiar base sequence. Or to
put it another way, the base sequence is not the sole intrinsic
determinant of mRNA function. Depending on location (within long
exons, around stop codons, within 3' UTRs), m6A
methylation has been implicated in RNA splicing and translational
control, and some m6A-binding factors that may contribute to these
processes have been identified” (Hastings 2013).
“The identification of m6A-responsive RNA-binding proteins
has revealed that m6A regulates cognate RNAs from nascence
to decay. In addition, m6A modulates RNA structure, miRNA
biology and protein localization. It remains unclear how these various
functions are coordinated within the cell, how they are coupled with
m6A biogenesis and removal, and how they are regulated in a
cell type- and cell state-dependent manner” (Liu and Pan 2016,
doi:10.1038/nsmb.3162).
“Cellular RNAs carry diverse chemical modifications that used to be
regarded as static and having minor roles in ‘fine-tuning’
structural and functional properties of RNAs. ... Recent studies
have discovered protein ‘writers’, ‘erasers’ and ‘readers’ of this
[m6A modification], as well as its dynamic deposition on
mRNA and other types of nuclear RNA. These findings strongly
indicate dynamic regulatory roles that are analogous to the
well-known reversible epigenetic modifications of DNA and histone
proteins. This reversible RNA methylation adds a new dimension to
the developing picture of post-transcriptional regulation of gene
expression” (Fu, Dominissini, Rechavi and He 2014).
“N6-adenosine methylation directs mRNAs to distinct fates by grouping
them for differential processing, translation and decay in processes
such as cell differentiation, embryonic development and stress
responses. Other mRNA modifications, including N1-methyladenosine,
5-methylcytosine and pseudouridine, together with m6A form the
epitranscriptome and collectively code a new layer of information that
controls protein synthesis”
(Zhao, Roundtree and He 2016, doi:10.1038/nrm.2016.132).
“Two studies in Nature report a role for m6A in the
regulation of sex determination in Drosophila melanogaster”.
“These studies provide important insights into the biogenesis and
function of m6A and raise the possibility that it has
widespread regulatory roles in development that may extend across
species” (Waldron 2017, doi:10.1038/nrm.2016.173).
“During the maternal-to-zygotic transition, maternal mRNAs are cleared
by multiple distinct but interrelated pathways. A recent study in
Nature by Zhao et al. (2017) finds that YTHDF2, a reader of
N6-methylation, facilitates maternal mRNA decay,
introducing an additional facet of control over transcript fate and
developmental reprogramming”
(Kontour and Giraldez 2017, doi:10.1016/j.devcel.2017.02.024).
“Studies from our group have shown that methylation of cellular 5'UTRs
is triggered by diverse stress-response pathways that are often
activated in human disease. We find that these 5'UTR m6A
residues promote the translation of stress-response proteins and
suggest the existence of a so-called m6A stress response,
in which m6A is a potential mechanism for fine-tuning the
production of critical proteins during disease states. Additional
physiological roles for m6A-mediated gene regulation that
have been uncovered include regulation of circadian rhythms, stem cell
differentiation, and noncoding RNA function. Additionally, several
studies have uncovered links between m6A regulation and
disease states: Multiple reports implicate various m6A
regulatory proteins in cancer, and several recent studies link
m6A to viral RNA stability”
(Meyer and Jaffrey 2017, doi:10.1146/annurev-cellbio-100616-060758).
New research “demonstrates that histone H3 lysine 36 trimethylation
(H3K36me3) facilitates co-transcriptional m6A modification
of mRNA ... [Analyses revealed that] approximately 70% of
m6A peaks overlapped with H3K36me3 sites and that the
overlapping sites were enriched near stop codons. Furthermore,
ectopic expression experiments suggested the link between the two
modifications was functional. Manipulations that lowered the levels of
H3K36me3 in cells also led to significantly lower global levels of
m6A, particularly around stop codons. By contrast, H3K36me3
was unaffected by reduced cellular levels of m6A. Taken
together, these observations suggest that H3K36me3 directs
m6A deposition, particularly at the 3ʹ end of coding
sequences ... observation suggests that H3K36me3 acts to recruit [a]
methyltransferase complex to RNA ... [indications are that]
m6A deposition occurs co-transcriptionally ... The
discovery of interplay between modified histones and RNA methylation
represents a new regulatory layer, and an additional level of
complexity, in the control of gene expression”
(Clyde 2019, doi:10.1038/s41576-019-0115-5).
“By using a cell fraction technique that separates
chromatin-associated nascent RNA, newly completed nucleoplasmic mRNA
and cytoplasmic mRNA, we have shown in a previous study that residues
in exons are methylated (m6A) in nascent pre-mRNA and
remain methylated in the same exonic residues in nucleoplasmic and
cytoplasmic mRNA. Thus, there is no evidence of a substantial degree
of demethylation in mRNA exons that would correspond to so-called
“epigenetic” demethylation. The turnover rate of mRNA molecules is
faster, depending on m6A content in HeLa cell mRNA,
suggesting that specification of mRNA stability may be the major role
of m6A exon modification. In mouse embryonic stem cells
(mESCs) lacking Mettl3, the major mRNA methylase, the cells continue
to grow, making the same mRNAs with unchanged splicing profiles in the
absence (>90%) of m6A in mRNA, suggesting no common
obligatory role of m6A in splicing. All these data argue
strongly against a commonly used “reversible dynamic
methylation/demethylation” of mRNA, calling into question the concept
of “RNA epigenetics” that parallels the well-established role of
dynamic DNA epigenetics”
(Darnell, Ke and Darnell 2018, doi:10.1261/rna.065219.117).
Response to previous item: “We agree with many of the viewpoints
expressed: that a majority of messenger RNA N6-methyladenosine
(m6A) methylation occurs cotranscriptionally, that one of
the main functions of m6A methylation on mRNA is to mark
sets of transcripts for expedited turnover, and that this methylation
may not dramatically affect splicing in HeLa cells. However, although
the impact of m6A methylation on splicing appears to be
modest in many cell lines, we suggest caution because m6A
methylation is enriched in long exons and overrepresented in
transcripts with alternative splicing variants (Dominissini et al.
2012). Several recent examples have revealed methylation-dependent
changes in splicing: One demonstrated m6A-modulated sex
determination in Drosophila melanogaster, another found
enhanced SAM synthetase expression mediated by a specific
m6A site installed by METTL16, and recent reports uncovered
extensive m6A-dependent splicing changes mediated by ALKBH5
in male germ lines, as well as FTO-involved pre-mRNA splicing changes.
The potential effects of RNA methylation on constitutive and
alternative splicing in additional physiological contexts need to be
further evaluated” (Zhao, Nachtergaele, Roundtree and He 2018,
doi:10.1261/rna.064295.117).
“Although the presence of m6A in an mRNA can affect its fate
in different ways, it is unclear how m6A directs this
process and why the effects of m6A can vary in different
cellular contexts. Here we show that the cytosolic
m6A-binding proteins—YTHDF1, YTHDF2 and YTHDF3—undergo
liquid–liquid phase separation in vitro and in cells. This phase
separation is markedly enhanced by mRNAs that contain multiple, but
not single, m6A residues. Polymethylated mRNAs act as a
multivalent scaffold for the binding of YTHDF proteins, juxtaposing
their low-complexity domains and thereby leading to phase separation.
The resulting mRNA–YTHDF complexes then partition into different
endogenous phase-separated compartments, such as P-bodies, stress
granules or neuronal RNA granules. m6A-mRNA is subject to
compartment-specific regulation, including a reduction in the
stability and translation of mRNA. These studies reveal that the
number and distribution of m6A sites in cellular mRNAs can
regulate and influence the composition of the phase-separated
transcriptome, and suggest that the cellular properties of
m6A-modified mRNAs are governed by liquid–liquid phase
separation principles”
(Ries, Zaccara, Klein et al. 2019, doi:10.1038/s41586-019-1374-1).
“Host cell metabolism can be modulated by viral infection, affecting
viral survival or clearance ... Here we report that in response to
viral infection, host cells impair the enzymatic activity of the RNA
m6A demethylase ALKBH5. This behavior increases the
m6A methylation on α-ketoglutarate dehydrogenase (OGDH)
messenger RNA (mRNA) to reduce its mRNA stability and protein
expression. Reduced OGDH decreases the production of the metabolite
itaconate that is required for viral replication. With reduced OGDH
and itaconate production in vivo, Alkbh5-deficient mice display innate
immune response–independent resistance to viral exposure”
(Liu, You, Lu et al. 2019, doi:10.1126/science.aax4468).
“The N6-methyladenosine (m6A) RNA modification is used
widely to alter the fate of mRNAs. Here we demonstrate that the C.
elegans writer METT-10 (the ortholog of mouse METTL16) deposits an
m6A mark on the 3′ splice site (AG) of the
S-adenosylmethionine (SAM) synthetase pre-mRNA, which inhibits its
proper splicing and protein production. The mechanism is triggered by
a rich diet and acts as an m6A-mediated switch to stop SAM
production and regulate its homeostasis. Although the mammalian SAM
synthetase pre-mRNA is not regulated via this mechanism, we show that
splicing inhibition by 3′ splice site m6A is conserved in
mammals. The modification functions by physically preventing the
essential splicing factor U2AF35 from recognizing the 3′ splice site”
(Mendel, Delaney, Pandey et al. 2021, doi:10.1016/j.cell.2021.03.062).
“m6A regulates chromatin state m6A is the most
abundant internal modification found on mRNA. This modification is
deposited cotranscriptionally by a methyltransferase complex at sites
bearing a specific sequence motif. This process is regulated by
transcription factors, modified histones and transcriptional rate.
Recent findings suggest that m6A-related pathways also
affect local transcriptiont. A new study by Li at al. shows that
co-transcriptional m6A modification of nascent mRNA
directly regulates the levels of dimethylated H3 K9 (H3K9me2) by
recruiting the demethylase KDM3B”
(Chan and Batista 2020, doi:10.1038/s41588-020-0685-3).
“Recent work by Zhang et al. has identified genomic sequence variants
that are associated with altered m6A levels in human
lymphoblastoid cell lines. As expected, the variants that
overlapped with m6A motif sites affected m6A
levels. Unexpectedly, however, variants overlapping with RBP-binding
sites near m6A motifs, as well as those modulating RNA
secondary structure, also influenced m6A levels. These
observations suggest that m6A levels are regulated by a
complex network of molecular processes that are not restricted to the
modification sites. This study also revealed that the effects of
m6A modification on molecular events leading to translation
are heterogeneous across transcripts and are affected by RBPs that
interact near the sites of modification. Additionally, some of the
sequence variants associated with altered m6A levels were
found at transcription-factor-binding sites. Remarkably, the effects
of variants in transcription-factor-binding sites on m6A
levels were comparable to the effects of variants in RNA features,
thus suggesting that the recruitment of the methyltransferase complex
may be mediated by transcription factors”
(Chan and Batista 2020, doi:10.1038/s41588-020-0685-3).
“The picture that emerges from these studies (see previous two bullet
items) is that of a complex system in which m6A levels are
targeted by multiple pathways in transcriptional regulation”
(Chan and Batista 2020, doi:10.1038/s41588-020-0685-3).
“Epigenetic modifications occur on genomic DNA and histones to
influence gene expression. More recently, the discovery that mRNA
undergoes similar chemical modifications that powerfully impact
transcript turnover and translation adds another layer of dynamic gene
regulation. Central to precise and synchronized regulation of gene
expression is intricate crosstalk between multiple checkpoints
involved in transcript biosynthesis and processing. There are more
than 100 internal modifications of RNA in mammalian cells. The most
common is N6-methyladenosine (m6A)
methylation. Although m6A is established to influence RNA
stability dynamics and translation efficiency, rapidly accumulating
evidence shows significant crosstalk between RNA methylation and
histone/DNA epigenetic mechanisms. These interactions specify
transcriptional outputs, translation, recruitment of chromatin
modifiers, as well as the deployment of the m6A
methyltransferase complex (MTC) at target sites. In this review, we
dissect m6A-orchestrated feedback circuits that regulate
histone modifications and the activity of regulatory RNAs, such as
long noncoding (lnc)RNA and chromosome-associated regulatory RNA.
Collectively, this body of evidence suggests that m6A acts
as a versatile checkpoint that can couple different layers of gene
regulation with one another”
(Kan, Chen and Sallam 2022, doi:10.1016/j.tig.2021.06.014).
“N6-methyladenosine or m6A modification to mRNAs is now
recognised as a key regulator of gene expression and protein
translation. The fate of m6A-modified mRNAs is decoded by
m6A readers, mostly found in the cytoplasm, except for the
nuclear-localised YTHDC1. While earlier studies have implicated
YTHDC1–m6A functions in alternative splicing and mRNA
export, recent literature has expanded its close association to the
chromatin-associated, noncoding and regulatory RNAs to fine-tune
transcription and gene expression in cells. Here, we summarise current
progress in the study of YTHDC1 function in cells, highlighting its
multiple modes of action in regulating gene expression, and propose
the formation of YTHDC1 nuclear condensates as a general mechanism
that underlies its diverse functions in the nucleus.
• “YTHDC1 interacts with m6A-modified RNAs to regulate multiple steps of RNA metabolism in the nucleus;
• “YTHDC1 is widely associated with transcriptional activation (via enhancer RNA-mediated crossregulation with active epigenetic marks);
• “YTHDC1 transcriptional repressive action is largely associated with transposable elements and long ncRNAs;
• “The diversity in YTHDC1–m6A functions is linked to their ability to promote membraneless nuclear subcompartments, such as the nuclear speckles”;
(Widagdo, Anggono and Wong 2022, doi:10.1016/j.tig.2021.11.005).
“Transcriptional regulation, which integrates chromatin accessibility,
transcription factors and epigenetic modifications, is crucial for
establishing and maintaining cell identity. The interplay between
different epigenetic modifications and its contribution to
transcriptional regulation remains elusive. Here, we show that
METTL3-mediated RNA N6-methyladenosine (m6A)
formation leads to DNA demethylation in nearby genomic loci in normal
and cancer cells, which is mediated by the interaction between
m6A reader FXR1 and DNA 5-methylcytosine dioxygenase TET1.
Upon recognizing RNA m6A, FXR1 recruits TET1 to genomic
loci to demethylate DNA, leading to reprogrammed chromatin
accessibility and gene transcription. Therefore, we have
characterized a regulatory mechanism of chromatin accessibility and
gene transcription mediated by RNA m6A formation coupled
with DNA demethylation, highlighting the importance of the crosstalk
between RNA m6A and DNA modification in physiologic and
pathogenic process”
(Deng, Zhang, Su et al. 2022, doi:10.1038/s41588-022-01173-1).
“We ... identify transposable element-associated proteins, and reveal
an interplay between RNA N6-methyladenosine
(m6A) and DNA methylation that is crucial for regulating TE
activation and human embryonic stem cell (hESC) fate”
(doi:10.1038/s41588-023-01453-4).
“Embryos across metazoan lineages can enter reversible states of
developmental pausing, or diapause, in response to adverse
environmental conditions ... Here we show that
N6-methyladenosine (m6A) RNA methylation by
Mettl3 is required for developmental pausing in mouse blastocysts and
embryonic stem (ES) cells. Mettl3 enforces transcriptional dormancy
through two interconnected mechanisms: (1) it promotes global mRNA
destabilization and (2) it suppresses global nascent transcription by
destabilizing the mRNA of the transcriptional amplifier and oncogene
N-Myc, which we identify as a crucial anti-pausing factor ... These
findings uncover Mettl3 as a key orchestrator of the crosstalk between
transcriptomic and epitranscriptomic regulation during developmental
pausing, with implications for dormancy in adult stem cells and
cancer”
(Collignon, Cho, Furlan et al. 2023, doi:10.1038/s41556-023-01212-x).
“Control of insulin mRNA translation is crucial for energy
homeostasis, but the mechanisms remain largely unknown. We discovered
that insulin mRNAs across invertebrates, vertebrates and mammals
feature the modified base N6-methyladenosine (m6A). In
flies, this RNA modification enhances insulin mRNA translation by
promoting the association of the transcript with polysomes. Depleting
m6A in Drosophila melanogaster insulin 2 mRNA (dilp2)
directly through specific 3′ untranslated region (UTR) mutations, or
indirectly by mutating the m6A writer Mettl3, decreases
dilp2 protein production, leading to aberrant energy homeostasis and
diabetic-like phenotypes. Together, our findings reveal adenosine
mRNA methylation as a key regulator of insulin protein synthesis with
notable implications for energy balance and metabolic disease”
(Wilinski and Dus 2023, doi:10.1038/s41594-023-01048-x).
“Previous studies reported an effect of N6-methyladenosine
(m6A) of super-enhancer RNAs (seRNAs) on chromatin
accessibility and gene transcription. We investigated seRNA
m6A levels in pancreatic ductal adenocarcinoma (PDAC) and
found that aberrantly increased m6A methylation promoted
local chromatin accessibility, resulting in increased transcription of
oncogenes acting in PDAC progression”
(doi:10.1038/s41588-023-01567-9).
- “In mammals, m6A occurs on average in 3–5 sites per mRNA molecule” (Pan 2013).
- m6A occurs at regions of mRNA highly conserved in a number of vertebrate species. Also, “the m6A modification exhibits tissue-specific regulation and is markedly increased throughout brain development. We find that m6A sites are enriched near stop codons and in 3' UTRs, and we uncover an association between m6A residues and microRNA-binding sites within 3' UTRs” A mammalian gene, FTO, has been found to demethylate N6-methyladenosine (m6A), and mutations that increase the activity of FTO are associated with elevated body mass index and increased risk for obesity (Meyer, Saletore, Zumbo et al. 2012).
- A rather dramatic finding (although the word “determines” reflects the usual sort of linguistic overreaching): “m6A methylation of RNA ... regulates RNA processing and determines the period and oscillatory stability of the mammalian circadian clockwork”. This effect is achieved in connection with a general function of m6A methylation, which “normally accelerates processing and nuclear export of RNA” (Hastings 2013, reporting on work by Fustin, Doi, Yamaguchi et al. 2013).
- An RNA-binding protein has been found that selectively recognizes and binds to m6A-containing RNA (noncoding RNA as well as mRNA). The protein can localize the RNA at RNA decay sites, and through its selective binding can affect the translation status and lifetime of the RNA. “We show that [the YTHDF2 protein] alters the distribution of the cytoplasmic states of several thousand m6A-containing mRNA. This present work demonstrates that reversible m6A deposition could dynamically tune the stability and localization of the target RNAs through m6A ‘readers’” (Wang, Lu, Gomez et al. 2014).
- “The mRNA targets of YTHDF2 contain many transcription factors, indicating that the m6A-dependent mRNA turnover could serve to dynamically adjust the expression of regulatory genes” (Wang, Zhao, Roundtree et al. 2015, doi:10.1016/j.cell.2015.05.014).
- “The stability of m6A-modified mRNA is regulated by an m6A reader protein, human YTHDF2, which recognizes m6A and reduces the stability of target transcripts. Looking at additional functional roles for the modification, we find that another m6A reader protein, human YTHDF1, actively promotes protein synthesis by interacting with translation machinery. In a unified mechanism of m6A-based regulation in the cytoplasm, YTHDF2-mediated degradation controls the lifetime of target transcripts, whereas YTHDF1-mediated translation promotion increases translation efficiency, ensuring effective protein production from dynamic transcripts that are marked by m6A. Therefore, the m6A modification in mRNA endows gene expression with fast responses and controllable protein production through these mechanisms (Wang, Zhao, Roundtree et al. 2015, doi:10.1016/j.cell.2015.05.014).
- “The regulatory role of N6-methyladenosine (m6A) and its nuclear binding protein YTHDC1 in pre-mRNA splicing remains an enigma. Here we show that YTHDC1 promotes exon inclusion in targeted mRNAs through recruiting pre-mRNA splicing factor SRSF3 (SRp20) while blocking SRSF10 (SRp38) mRNA binding ... [Experimental work showed] that YTHDC1-regulated exon-inclusion patterns were similar to those of SRSF3 but opposite of SRSF10 [and that there is] a competitive binding of SRSF3 and SRSF10 to YTHDC1. Moreover, YTHDC1 facilitates SRSF3 but represses SRSF10 in their nuclear speckle localization, RNA-binding affinity, and associated splicing events, dysregulation of which, as the result of YTHDC1 depletion, can be restored by reconstitution with wild-type, but not m6A-binding-defective, YTHDC1. Our findings provide the direct evidence that m6A reader YTHDC1 regulates mRNA splicing through recruiting and modulating pre-mRNA splicing factors for their access to the binding regions of targeted mRNAs” (Xiao, Adhikari, Dahals et al. 2016, doi:10.1016/j.molcel.2016.01.012).
- “N6-methyladenosine (m6A) residues within the 5' UTR of mRNAs promote translation initiation through a mechanism that does not require the 5' cap or cap-binding proteins. Diverse cellular stresses selectively increase the levels of m6A within 5' UTRs, suggesting that 5' UTR m6A is important for mediating stress-induced translational responses” (blurb in Cell for article by Mitchell and Parker 2015, doi:10.1016/j.cell.2015.10.056).
- How might m6A methylation mediate such regulatory processes? It turns out that m6A can alter mRNA and lncRNA secondary structure, exposing otherwise hidden binding sites for RNA-binding proteins. “Here we show in human cells that m6A controls the RNA-structure-dependent accessibility of RNA binding motifs to affect RNA–protein interactions for biological regulation; we term this mechanism ‘the m6A-switch’”. The so-called “switch” was found to regulate the abundance and alternative splicing of target mRNAs (Liu, Dai, Zheng et al. 2015).
- “We identify m6A mRNA methylation as a regulator acting at molecular switches, during resolution of murine naïve pluripotency, to safeguard an authentic and timely down-regulation of pluripotency factors, which is needed for proper lineage priming and differentiation” (Geula, Moshitch-Moshkovitz, Dominissini et al. 2015; doi:10.1126/science.1261417).
- Reporting on work by Wang, Li, Toth et al. (2014): “Together, the data indicate that the pluripotency of embryonic stem cells is maintained, in part, through an RNA regulatory mechanism involving the m6A modification of developmental regulators, which blocks HUR binding, increases RISC binding and decreases mRNA stability to decrease gene expression. As thousands of mammalian mRNAs and long non-coding RNAs show m6A modification, this mechanism might have more widespread implications in various cell types” (Minton 2014).
- Questions. “Several fundamental questions remain. Prime among these is how specificity of modification is achieved. Clearly, the sequence that constitutes the consensus site is not sufficient on its own, nor does secondary structure appear to play a role. If methylation is cotranscriptional, it may be possible that chromatin status could play a role in site selection. The function(s) of m6A in nuclear RNA metabolism are also unclear. Although it is possible that nuclear factors recognize the modification, it seems equally plausible that modifications could function by preventing or altering the binding of some proteins. The importance or function of modifications in the vicinity of stop codons remains to be established, as does the importance of the FTO demethylase. Perhaps the most challenging question — and most difficult to answer — is how such a widespread modification has apparently quite specific effects. Are all modified sites equally important, or is only a small subset of them important?” (Nilsen 2014).
- “We demonstrate that m6A modification of mRNAs is co-transcriptional and depends upon the dynamics of the transcribing RNAPII. Suboptimal transcription rates lead to elevated m6A content, which may result in reduced translation. This study uncovers a general and widespread link between transcription and translation that is governed by epigenetic modification of mRNAs” (Slobodin, Han, Calderone et al. 2017, doi:10.1016/j.cell.2017.03.031).
- “What does become clear ... is that m6A deposition plays essential roles in mRNA metabolism, and both m6A methylases and demethylases are crucial during embryonic development and homeostasis of the central nervous, cardiovascular and reproductive systems. Furthermore, aberrant m6A methylation pathways are linked to a range of human diseases including infertility, obesity as well as developmental and neurological disorders” (Blanco and Frye 2014, doi:10.1016/j.ceb.2014.06.006).
- “we only described current advances on m5C and m6A methylation, but a large number of other intriguing chemical modifications exist in RNAs. Thus, our current knowledge only scratches the surface of the many roles of post-transcriptional modifications in modulating transcriptional and translational processes” (Blanco and Frye 2014, doi:10.1016/j.ceb.2014.06.006).
- “Here we show that in response to heat shock stress, certain adenosines within the 5' UTR of newly transcribed mRNAs are preferentially methylated. We find that the dynamic 5' UTR methylation is a result of stress-induced nuclear localization of YTHDF2, a well-characterized m6A ‘reader’. Upon heat shock stress, the nuclear YTHDF2 preserves 5' UTR methylation of stress-induced transcripts by limiting the m6A ‘eraser’ FTO from demethylation. Remarkably, the increased 5' UTR methylation in the form of m6A promotes cap-independent translation initiation, providing a mechanism for selective mRNA translation under heat shock stress. Using Hsp70 mRNA as an example, we demonstrate that a single m6A modification site in the 5' UTR enables translation initiation independent of the 5' end N7-methylguanosine cap. The elucidation of the dynamic features of 5' UTR methylation and its critical role in cap-independent translation not only expands the breadth of physiological roles of m6A, but also uncovers a previously unappreciated translational control mechanism in heat shock response” (Zhou, Wan, Gao et al. 2015, doi:10.1038/nature15377).
- “The long non-coding RNA X-inactive specific transcript (XIST) mediates the transcriptional silencing of genes on the X chromosome. Here we show that, in human cells, XIST is highly methylated with at least 78 N6-methyladenosine (m6A) residues ... We show that m6A formation in XIST, as well as in cellular mRNAs, is mediated by RNA-binding motif protein 15 (RBM15) and its paralogue RBM15B, which bind the m6A-methylation complex and recruit it to specific sites in RNA. This results in the methylation of adenosine nucleotides in adjacent m6A consensus motifs. Furthermore, we show that knockdown of RBM15 and RBM15B, or knockdown of methyltransferase like 3 (METTL3), an m6A methyltransferase, impairs XIST-mediated gene silencing. A systematic comparison of m6A-binding proteins shows that YTH domain containing 1 (YTHDC1) preferentially recognizes m6A residues on XIST and is required for XIST function” (Patil, Chen, Pickering et al. 2016, doi:10.1038/nature19342).
- “The m6A demethylation activity of ALKBH5 critically impacts mRNA nuclear export and spermatogenesis, and both enzymes participate in the various disease mechanisms related to cancer. A recent study discovered that the [methyltransferase] METTL3-METTL14 complex is rapidly recruited to the DNA damage site created by UV irradiation, where it mediates local RNA m6A methylation. This process facilitates recruitment of DNA damage repair polymerase κ and can be reversed by [the demethylase] FTO within a short period of time. These studies are building a framework for understanding how methyltransferases and demethylases actively control methylation dynamics in homeostatic and acute responses to cellular stimuli” (Roundtree, Evans, Pan and He 2017, doi:10.1016/j.cell.2017.05.045).
- “The position of [the m6] base modification affects its impact on translation. Under most conditions examined thus far, m6A is seen to be enriched near 3' ends of mRNA ORFs and 3' UTRs, where it has been shown to affect mRNA stability, translation, and polyA site choice. Under heat shock and other cellular stress conditions, there is removal of 3' m6A modifications and a relative increase in 5' leader m6A modifications for a subset of messages, including that for the chaperone Hsp70. These new 5' m6A bases are capable of directly recruiting the translation initiation factor eIF3 and enabling translation initiation independent of the canonical eIF4E cap-binding complex. It is interesting to note that, in contrast to the canonical cap-dependent translation initiation model, which requires eIF4E-cap interaction, recent work has shown that eIF3 can form an alternate cap-binding complex. This result suggests that pervasive perception of ‘cap-dependent’ as generally synonymous with eIF4E-dependent may require revisiting, and that choice between these two types of cap-dependent initiation may represent a broad new mode of selective translation initiation control. The eIF4E-independent translation initiation seen for Hsp70 is highly specific to m6A modifications and also highly dependent on the context of the modification. A non-structured 5' mRNA end is required, implying a scanning rather than IRES-based mechanism of translation initiation, and the effect was seen to be strongest when the methylated A was flanked by a 5' G and 3' C base. m6A modifications outside of the 5' leader did not retain this activity. Given the reversibility of this modification, these results suggest a mechanism by which mRNA [translation] initiation cues can be dynamically and selectively modulated” (Brar 2016, doi:10.1016/j.cell.2016.09.022).
- “tRNAs contain the largest number of modifications with the widest chemical diversity. Eukaryotic tRNAs contain on average 13 modifications per molecule ranging from base isomerization and base and ribose methylations to elaborate addition of ring structures. tRNA modifications contribute to the efficiency and fidelity of decoding, as well as folding, cellular stability, and localization. Human rRNA contains >210 modification sites including 2'-O-methyls, pseudouridines, and base methylations. Ribosomal RNAs present a striking example of how chemical modifications support functions as, without internal pseudouridines and 2'-O-methylated sugars, rRNA biogenesis is blocked. Human spliceosomal RNAs contain >50 modification sites including 2'-O-methyls, pseudouridines, and base methylations. Some of these modifications are known to be important in the RNA splicing reaction. (Roundtree, Evans, Pan and He 2017, doi:10.1016/j.cell.2017.05.045).
- “Here we report on a new mRNA modification, N1-methyladenosine (m1A), that occurs on thousands of different gene transcripts in eukaryotic cells, from yeast to mammals, at an estimated average transcript stoichiometry of 20% in humans. We show that m1A is enriched around the start codon upstream of the first splice site: it preferentially decorates more structured regions around canonical and alternative translation initiation sites, is dynamic in response to physiological conditions, and correlates positively with protein production. These unique features are highly conserved in mouse and human cells, strongly indicating a functional role for m1A in promoting translation of methylated mRNA” (Dominissini, Nachtergaele, Moshitch-Moshkovitz et al. 2016, doi:10.1038/nature16998).
- “In analogy to the regulation of gene expression by miRNAs, we propose that the main function of m6A is post-transcriptional fine-tuning of gene expression. In contrast to miRNA regulation, which mostly reduces gene expression, we argue that m6A provides a fast mean to post-transcriptionally maximize gene expression. Additionally, m6A appears to have a second function during developmental transitions by targeting m6A-marked transcripts for degradation” (Roignant and Soller 2017, doi:10.1016/j.tig.2017.04.003).
- “YTH-domain proteins can specifically recognize m6A modification to control mRNA maturation, translation and decay. m6A can also alter RNA structures to affect RNA–protein interactions in cells. Here, we show that m6A increases the accessibility of its surrounding RNA sequence to bind heterogeneous nuclear ribonucleoprotein G (HNRNPG). Furthermore, HNRNPG binds m6A-methylated RNAs through its C-terminal low-complexity region, which self-assembles into large particles in vitro. The Arg-Gly-Gly repeats within the low-complexity region are required for binding to the RNA motif exposed by m6A methylation. We identified 13,191 m6A sites in the transcriptome that regulate RNA–HNRNPG interaction and thereby alter the expression and alternative splicing pattern of target mRNAs. Low-complexity regions are pervasive among mRNA binding proteins. Our results show that m6A-dependent RNA structural alterations can promote direct binding of m6A-modified RNAs to low-complexity regions in RNA binding proteins” (Liu, Zhou, Parisien et al. 2017, doi:10.1093/nar/gkx141).
- “we show that [proteins involved in miRNA biogenesis] Dgcr8 and Drosha physically associate with chromatin in murine embryonic stem cells, specifically with a subset of transcribed coding and noncoding genes. Dgcr8 recruitment to chromatin is dependent on transcription as well as methyltransferase-like 3 (Mettl3), which catalyzes RNA N6-methyladenosine (m6A). Intriguingly, we found that acute temperature stress causes radical relocalization of Dgcr8 and Mettl3 to heat-shock genes, where they act to co-transcriptionally mark mRNAs for subsequent RNA degradation. Together, our findings elucidate a novel mode of co-transcriptional gene regulation, in which m6A serves as a chemical mark that instigates subsequent post-transcriptional RNA-processing events” (Knuckles, Carl, Musheev et al. 2017, doi:10.1038/nsmb.3419).
- “m6A modification is catalysed by METTL3 and enriched in the 3′ untranslated region of a large subset of mRNAs at sites close to the stop codon. METTL3 can promote translation but the mechanism and relevance of this process remain unknown. Here we show that METTL3 enhances translation only when tethered to reporter mRNA at sites close to the stop codon, supporting a mechanism of mRNA looping for ribosome recycling and translational control ... We identify a direct physical and functional interaction between METTL3 and the eukaryotic translation initiation factor 3 subunit h (eIF3h). METTL3 promotes translation of a large subset of oncogenic mRNAs — including bromodomain-containing protein 4 — that is also m6A-modified in human primary lung tumours. The METTL3–eIF3h interaction is required for enhanced translation, formation of densely packed polyribosomes and oncogenic transformation” (Choe, Lin, Zhang et al. 2018, doi:10.1038/s41586-018-0538-8).
- “RNA methylation provides a protective effect in maintaining cellular integrity by clearing reactive endogenous retrovirus-derived RNA species, which may be especially important when transcriptional silencing is less stringent” (Chelmicki, Roger, Teissandier et al. 2021, doi:10.1038/s41586-020-03135-1)
-
mRNA adenosine methylation (m6Am)
“Mauer et al. report that one of the most prevalent modified
bases, N6,2ʹ- O-dimethyladenosine
(m6Am), found in 30% of mRNAs, is a
dynamic and reversible modification that confers mRNA stability. In
contrast to internal base modifications such as N6-methyladenosine
(m6A), m6Am is found at the 5ʹ end of
mRNAs, when the first nucleotide following the 5ʹ cap is a
2ʹ-O-methyladenosine that is modified by additional
N6-methylation.” This reversible modification protects
against mRNA decapping and degradation, and leads to increased mRNA
transcription. “The preference of [the demethylase] FTO for
m6Am also raises doubts over some of the
previously established dynamics of the m6A modification”
(Koch 2016, doi:10.1038/nrg.2016.165).
-
mRNA guanosine methylation
Blurb in Science for doi:10.1016/j.molcel.2018.06.001 (2018):
“Transfer RNAs (tRNAs), the adaptor molecules between messenger RNAs
(mRNAs) and ribosomes during translation, are subjected to various
types of chemical modifications, one of which is
N7-methyguanosine (m7G). Mutations in the human
m7G methyltransferase complex lead to developmental
disorders such as microcephalic primordial dwarfism and Down syndrome.
Lin et al. mapped the m7G tRNA methylome at
single-nucleotide resolution and demonstrated its essential role in
mouse embryonic stem cells. Depletion of members of the m7G
methyltransferase complex resulted in increased ribosome pausing on,
and inefficient translation of, mRNAs involved in the cell cycle and
brain development, thereby disrupting differentiation to neural
lineages. This study is an important step toward a fuller
understanding of how defects in tRNA methylation cause
neurodevelopmental disorders.”
-
mRNA cytosine methylation
mRNA cytosine methylation involves the “decoration” of RNA cytosine
bases with a methyl group. Little is known about the functional
implications of this modification, but given the research
techniques available today, together with numerous tantalizing
clues, the field seems poised for major discoveries (Motorin, Lyko
and Helm 2010; Squires and Preiss 2010).
- “Surprisingly, we discovered 10,275 [methylated cytosine] sites in [human] mRNAs and ... non-coding RNAs. We observed that distribution of modified cytosines between RNA types was not random; within mRNAs they were enriched in the untranslated regions and near Argonaute binding regions. ... Our data demonstrates the widespread presence of modified cytosines throughout coding and non-coding sequences in a transcriptome, suggesting a broader role of this modification in the post-transcriptional control of cellular RNA function” (Squires, Patel and Nousch 2012).
- An enzyme that performs methylation of tRNA and mRNA cytosines (NSUN2) varies in its localization within the cell (nucleolus versus cytoplasm) during the cell cycle, and is itself regulated by phosphorylation (Squires, Patel and Nousch 2012).
- “Although 5-methylcytosine (m5C) is a widespread modification in RNAs, its regulation and biological role in pathological conditions (such as cancer) remain unknown. Here, we provide the single-nucleotide resolution landscape of messenger RNA m5C modifications in human urothelial carcinoma of the bladder (UCB). We identify numerous oncogene RNAs with hypermethylated m5C sites causally linked to their upregulation in UCBs and further demonstrate YBX1 as an m5C ‘reader’ recognizing m5C-modified mRNAs ... YBX1 maintains the stability of its target mRNA [thereby supporting cancerous growth] by recruiting ELAVL1. Moreover, NSUN2 and YBX1 are demonstrated to drive UCB pathogenesis by [methylating the RNA in the 3′ untranslated region]”
- “Modified nucleotides within cellular RNAs significantly influence their biogenesis, stability, and function. As reviewed here, 3-methylcytidine (m3C) has recently come to the fore through the identification of the methyltransferases responsible for installing m3C32 in human tRNAs. Mechanistic details of how m3C32 methyltransferases recognize their substrate tRNAs have been uncovered and the biogenetic and functional relevance of interconnections between m3C32 and modified adenosines at position 37 highlighted. Functional insights into the role of m3C32 modifications indicate that they influence tRNA structure and, consistently, lack of m3C32 modifications impairs translation. Development of quantitative, transcriptome-wide m3C mapping approaches and the discovery of an m3C demethylase reveal m3C to be dynamic, raising the possibility that it contributes to fine-tuning gene expression in different conditions (Bohnsack, Kleiber, Lemus-Diaz and Bohnsack 2022, doi:10.1016/j.tibs.2022.03.004).
-
mRNA cytosine hydroxymethylation
- “Hydroxymethylcytosine, well described in DNA, occurs also in RNA. Here, we show that hydroxymethylcytosine preferentially marks polyadenylated RNAs and is deposited by Tet in Drosophila. We map the transcriptome-wide hydroxymethylation landscape, revealing hydroxymethylcytosine in the transcripts of many genes, notably in coding sequences, and identify consensus sites for hydroxymethylation. We found that RNA hydroxymethylation can favor mRNA translation. Tet and hydroxymethylated RNA are found to be most abundant in the Drosophila brain, and Tet-deficient fruitflies suffer impaired brain development, accompanied by decreased RNA hydroxymethylation” (Delatte, Wang, Ngoc et al. 2016, doi:10.1126/science.aac5253).
-
mRNA cytidine acetylation
Now acetylated cytidine — N4-acetylcytidine
(ac4C) — has been identified “as a widespread mark in
cellular mRNAs that influences both mRNA stability and translation”.
This modification renders translation more efficient “by promoting
efficient tRNA decoding”. The acetylation is “carried out by
N-acetyltransferase 10 (NAT10), which possesses both acetyltransferase
and RNA binding domains”. “Many of the acetylated transcripts ... are
important for cell survival and viability. This is consistent with
previous studies indicating that NAT10 affects the cellular growth
rate, as well as with the authors’ discovery that NAT10 −/− cells
exhibit decreased proliferation”
(Choi and Meyer 2018, doi:10.1038/s41594-018-0159-9).
-
Small RNA glycosylation
“Glycans modify lipids and proteins to mediate inter- and
intramolecular interactions across all domains of life. RNA is not
thought to be a major target of glycosylation. Here, we challenge this
view with evidence that mammals use RNA as a third scaffold for
glycosylation. Using a battery of chemical and biochemical approaches,
we found that conserved small noncoding RNAs bear sialylated glycans.
These “glycoRNAs” were present in multiple cell types and mammalian
species, in cultured cells, and in vivo. GlycoRNA assembly
depends on canonical N-glycan biosynthetic machinery and results in
structures enriched in sialic acid and fucose. Analysis of living
cells revealed that the majority of glycoRNAs were present on the cell
surface and can interact with anti-dsRNA antibodies and members of the
Siglec receptor family. Collectively, these findings suggest the
existence of a direct interface between RNA biology and glycobiology,
and an expanded role for RNA in extracellular biology”
(Flynn, Pedram, Malaker et al. 2021, doi:10.1016/j.cell.2021.04.023).
-
tRNA and rRNA modifications
“Tens of millions of tRNA transcripts are present in a human cell, and
tRNA is the most extensively modified RNA in a cell. The
modifications are highly diverse, and their functions depend on the
location within a tRNA and its chemical nature. The most common tRNA
molecules consist of 76 nucleotides 10 . A human tRNA molecule, on
average, contains 11–13 different modifications. Accordingly, a large
number of enzymes are involved in the site-specific deposition of the
modifications. The modifications range from simple methylation or
isomerisation ... to complex multistep chemical modifications”
(Delaunay and Frye 2019, doi:10.1038/s41556-019-0319-0).
“Ribosomal RNA (rRNA) is the most abundant type of RNA in a cell.
Around 130 individual rRNA modifications have recently been visualized
in the three-dimensional structure of the human 80S ribosome. The most
abundant rRNA modifications in eukaryotes are 2′-O-methylation of the
ribose and the isomerisation of uridine to pseudouridine (Ψ). Most
rRNA modifications occur in or close to functionally important sites
and can facilitate efficient and accurate protein synthesis when they
occur — for instance, at the peptidyltransferase center and the
decoding site”
(Delaunay and Frye 2019, doi:10.1038/s41556-019-0319-0).
“Post-transcriptional modification of tRNA is universally required
for accurate and efficient translation. Modifications are found in
all characterized tRNA species, and are highly conserved within
each domain of life. Modifications have a number of different
roles, with well documented examples including modulating the
efficiency and specificity of charging, altering the specificity of
decoding, maintaining the frame for decoding, and preventing decay
of pre-tRNA and mature tRNA”.
“Post-transcriptional tRNA modifications are critical for efficient and accurate translation, and have multiple different roles. Lack of modifications often leads to different biological consequences in different organisms, and in humans is frequently associated with neurological disorders” (Guy and Phizicky 2014).
“tRNA research is blooming again, with demonstration of the
involvement of tRNAs in various other pathways beyond translation and
in adapting translation to environmental cues. These roles are linked
to the presence of tRNA sequence variants known as isoacceptors and
isodecoders, various tRNA base modifications, the versatility of
protein binding partners and tRNA fragmentation events, all of which
collectively create an incalculable complexity. This complexity
provides a vast repertoire of tRNA species that can serve various
functions in cellular homeostasis and in adaptation of cellular
functions to changing environments”. “Fragmentation repurposes tRNAs
to functions outside of translation, including regulation of gene
expression and epigenetics”
(Schimmel 2018, doi:10.1038/nrm.2017.77).
“In response to environmental cues, tRNA modifications can act as a
rheostat of protein synthesis rates via at least two mechanisms.
First, modifications outside the anticodon loop often modulate the
rate of global de novo protein synthesis mostly through regulating
tRNA biogenesis. Second, modifications within the anticodon loop can
determine the translation speed of codon-specific genes. Because
wobble base modifications usually affect gene-specific translation,
they have the potential to directly modulate distinct cellular
functions such as survival, growth and differentiation”
(Delaunay and Frye 2019, doi:10.1038/s41556-019-0319-0).
“A wide variety of tRNA modifications are found in the tRNA anticodon,
which are crucial for precise codon recognition and reading frame
maintenance, thereby ensuring accurate and efficient protein
synthesis. In addition, tRNA-body regions are also frequently modified
and thus stabilized in the cell. Over the past two decades, 16 novel
tRNA modifications were discovered in various organisms, and the
chemical space of tRNA modification continues to expand. Recent
studies have revealed that tRNA modifications can be dynamically
altered in response to levels of cellular metabolites and
environmental stresses. Importantly, we now understand that
deficiencies in tRNA modification can have pathological consequences,
which are termed ‘RNA modopathies’. Dysregulation of tRNA modification
is involved in mitochondrial diseases, neurological disorders and
cancer” (Suzuki 2021, doi:10.1038/s41580-021-00342-0).
“Until now, enzymatic phosphorylation of RNA had been observed only at
the molecule’s ends. Writing in Nature, Ohira et al. report the
internal phosphorylation of transfer RNA (tRNA) — a nucleic acid
responsible for translating messenger RNA into protein. The authors’
comprehensive study reveals that the properties of tRNA change after
internal phosphorylation, improving its ability to participate in
protein synthesis at high temperatures”
(Helm and Motorin 2022, doi:10.1038/d41586-022-01021-6).
- One research team reports results that suggest the “widespread importance of 2'-O-methylation of the tRNA anticodon loop, implicate tRNAPhe [the tRNA associated with the amino acid, phenylalanine] as the crucial substrate, and suggest that this modification circuitry is important for human neuronal development”. Working with yeast, the researchers also provide evidence “indicating that levels of tRNA modifications are regulated by cellular growth conditions” (Guy and Phizicky 2014, doi:10.1261/rna.047639.114.).
-
Alternative cleavage, polyadenylation, and deadenylation
[This section should be combined with Alternative coding sequences (transcription start and termination), above, to make a single large section entitled “Alternative transcriptional start and end processing”, or something like that. The contents of these two sections tend to bridge “decision-making during transcription” and “post-transcriptional decision-making”. Also, deadenylation should be treated under “RNA degradation” in the “post-transcriptional decision-making” section.]
Polyadenylation is the addition of a “tail” (consisting of multiple
adenosine monophosphates) to a nascent mRNA molecule. In mammals,
70-79% of mRNA molecules are thought to have more than one site where
they may be cleaved during transcription and then have a tail added.
(A recent estimate for humans is 50%.) This is known as “alternative
polyadenylation”.
“Alternative polyadenylation (APA) generates mRNAs with varying 3'
termini. It is regulated by variation in the concentration of cleavage
and polyadenylation factors and by RNA-binding proteins, as well as by
splicing and transcription. APA is important for cell proliferation and
differentiation owing to its roles in mRNA metabolism and protein
diversification”
(TOC blurb for Tian and Manley 2016, doi:10.1038/nrm.2016.116).
“Formation of the 3′ end of a eukaryotic mRNA is a key step in the
production of a mature transcript. This process is mediated by a number
of protein factors that cleave the pre-mRNA, add a poly(A) tail, and
regulate transcription by protein dephosphorylation. Cleavage and
polyadenylation specificity factor (CPSF) in humans, or cleavage and
polyadenylation factor (CPF) in yeast, coordinates these enzymatic
activities with each other, with RNA recognition, and with transcription.
The site of pre-mRNA cleavage can strongly influence the translation,
stability, and localization of the mRNA. Hence, cleavage site selection
is highly regulated”
(Boreikaitė and Passmore 2023; doi:10.1146/annurev-biochem-052521-012445).
Most polyadenylation sites are located within the 3' UTR.
“As 3' UTRs contain cis elements that are involved in various
aspects of mRNA metabolism, 3' UTR-APA can considerably affect
post-transcriptional gene regulation in various ways, including through
the modulation of mRNA stability, translation, nuclear export and
cellular localization, and even through effects on the localization of
the encoded protein. One remarkable feature of 3' UTR-APA is that it can
be regulated globally, simultaneously involving numerous transcripts in a
cell” (Tian and Manley 2016, doi:10.1038/nrm.2016.116).
“Alternative polyadenylation patterns are, to a great extent, tissue
specific”
(Tian and Manley 2016, doi:10.1038/nrm.2016.116).
It appears, contrary to previous thought, that “abundant and efficiently
translated mRNAs tend to have short poly(A) tails”. A study of
roundworms focused on how polyadenylate-binding proteins (PABPs) bind
poly(A) tails and either increase or reduce mRNA stability and
translation. “The authors found that the most abundant species of
polyadenylated mRNAs had poly(A) tails of 33–34 nucleotides (nt), which
is similar to the PABP footprint of 25–30 nt. They also saw a sharp drop
in abundance of mRNAs with tails shorter than 30 nt and found that tail
lengths were not distributed evenly but in increments of ∼30 nt, which is
indicative of serial binding of PABPs. This suggested that 3′ adenosines
not protected by PABP binding are removed and that the minimal tail
length required for transcript stability is that which is covered by a
single PABP.
“Further analysis revealed that mRNA species with shorter median poly(A) tail lengths were, on average, much more abundant than those with longer tails ... transcripts that were translationally activated during larval development had a significantly shorter median poly(A) tail size compared with those that were translationally repressed. Importantly, the correlation between high mRNA and translation levels and short poly(A) tails was found to be conserved in other eukaryotes.
“Interestingly, almost all genes produced transcripts with very long (>200 nt) poly(A) tails, indicating that well-expressed mRNAs undergo controlled poly(A) tail shortening (pruning). In support of this, tails of the majority of highly expressed and codon-optimized genes had lengths that would accommodate one or two PABPs (∼30–60 nt), whereas less-abundant mRNAs with poorly optimized codons had much longer poly(A) tails and a wider distribution of lengths” (Zlotorynski 2018, doi:10.1038/nrm.2017.120).
“The poly(A) tail of mRNA has been thought to be a pure stretch of
adenosine nucleotides with little informational content except for
length. Lim et al. identified enzymes that can decorate poly(A) tails
with non-A nucleotides. The noncanonical poly(A) polymerases, TENT4A and
TENT4B, incorporate intermittent non-A residues (G, U, or C) with a
preference for guanosine, which results in a heterogenous poly(A) tail.
Deadenylases trim poly(A) tails to initiate mRNA degradation but stall at
the non-A residues. In effect, the not-so-pure tail stabilizes mRNAs by
slowing down deadenylation.”
(Lim, Kim, Lee et al. 2018, 10.1126/science.aam5794)
“We find that the [polyadenylation] sequence can modulate gene expression
by over five orders of magnitude”
(Slutskin, Weinberger and Segal 2019, doi:10.1101/gr.247312.118).
“Premature transcription termination [PTT] is widespread in metazoans. It
can occur close to the transcription start site or further downstream in
the gene body. PTT generates transcripts that, depending on the
circumstances, are either rapidly degraded, or are stabilised by
polyadenylation, thus contributing to transcriptome diversification.
Stable premature transcripts can have independent functions as noncoding
(nc)RNA or mRNA encoding proteins with different properties compared with
those generated by the full-length transcript. PTT can negatively
regulate expression of the full-length transcript and especially controls
genes encoding transcriptional regulators. Factors triggering PTT
include not only canonical RNA 3′ processing and termination factors, but
also other players. Many metazoan factors oppose PTT, thus limiting its
damaging potential”
(Kamieniarz-Gdula and Proudfoot 2019, doi:10.1016/j.tig.2019.05.005).
“Analogous to alternative splicing, alternative polyadenylation (APA) has
long been thought to occur independently at proximal and distal polyA
sites ... we unexpectedly identified several hundred APA genes in human
cells whose distal polyA isoforms are retained in chromatin/nuclear
matrix and whose proximal polyA isoforms are released into the cytoplasm
... [We found] evidence that the strong distal polyA sites are processed
first and the resulting transcripts are subsequently anchored in
chromatin/nuclear matrix to serve as precursors for further processing at
proximal polyA sites ... Therefore, unlike alternative splicing, APA
sites are recognized independently, and in many cases, in a sequential
manner. This provides a versatile strategy to regulate gene expression in
mammalian cells”
(Tang, Yang, Li et al. 2022, doi:10.1038/s41594-021-00709-z).
“In eukaryotes, poly(A) tails are present on almost every mRNA. Early
experiments led to the hypothesis that poly(A) tails and the cytoplasmic
polyadenylate-binding protein (PABPC) promote translation and prevent
mRNA degradation, but the details remained unclear. More recent data
suggest that the role of poly(A) tails is much more complex:
poly(A)-binding protein can stimulate poly(A) tail removal
(deadenylation) and the poly(A) tails of stable, highly translated mRNAs
at steady state are much shorter than expected. Furthermore, the rate of
translation elongation affects deadenylation. Consequently, the interplay
between poly(A) tails, PABPC, translation and mRNA decay has a major role
in gene regulation. In this Review, we discuss recent work that is
revolutionizing our understanding of the roles of poly(A) tails in the
cytoplasm. Specifically, we discuss the roles of poly(A) tails in
translation and control of mRNA stability and how poly(A) tails are
removed by exonucleases (deadenylases), including CCR4–NOT and PAN2–PAN3”
(Passmore and Coller 2022, doi:10.1038/s41580-021-00417-y).
“Alternative polyadenylation (APA) generates transcript isoforms that
differ in the position of the 3′ cleavage site, resulting in the
production of mRNA isoforms with different length 3′ UTRs ... We
identified >500 Drosophila genes that express mRNA isoforms with a long
3′ UTR in proliferating spermatogonia but a short 3′ UTR in
differentiating spermatocytes due to APA. We show that the stage-specific
choice of the 3′ end cleavage site can be regulated by the arrangement of
a canonical polyadenylation signal (PAS) near the distal cleavage site
but a variant or no recognizable PAS near the proximal cleavage site. The
emergence of transcripts with shorter 3′ UTRs in differentiating cells
correlated with changes in expression of the encoded proteins, either
from off in spermatogonia to on in spermatocytes or vice versa. Polysome
gradient fractionation revealed >250 genes where the long 3′ UTR versus
short 3′ UTR mRNA isoforms migrated differently, consistent with dramatic
stage-specific changes in translation state. Thus, the developmentally
regulated choice of an alternative site at which to make the 3′ end cut
that terminates nascent transcripts can profoundly affect the suite of
proteins expressed as cells advance through sequential steps in a
differentiation lineage”
(Berry, Olivares, Gallicchio et al. 2022, doi:10.1101/gad.349689.122).
“Alternative cleavage and polyadenylation (APA) is a widespread mechanism
to generate mRNA isoforms with alternative 3′ untranslated regions
(UTRs). The expression of alternative 3′ UTR isoforms is highly cell type
specific and is further controlled in a gene-specific manner by
environmental cues. In this Review, we discuss how the dynamic,
fine-grained regulation of APA is accomplished by several mechanisms,
including cis-regulatory elements in RNA and DNA and factors that control
transcription, pre-mRNA cleavage and post-transcriptional processes.
Furthermore, signalling pathways modulate the activity of these factors
and integrate APA into gene regulatory programmes. Dysregulation of APA
can reprogramme the outcome of signalling pathways and thus can control
cellular responses to environmental changes. In addition to the
regulation of protein abundance, APA has emerged as a major regulator of
mRNA localization and the spatial organization of protein synthesis. This
role enables the regulation of protein function through the addition of
post-translational modifications or the formation of protein–protein
interactions”
(Mitscka and Mayr 2022, doi:10.1038/s41580-022-00507-5).
- Alternative cleavage sites and polyadenylation result in differing 3'-UTR lengths for the same mRNA. One result of this for mRNAs encoding membrane proteins is a change in localization of the protein. “The long 3' UTR of CD47 [a transmembrane protein also known as ‘integrin associated protein’] enables efficient cell surface expression of CD47 protein, whereas the short 3' UTR primarily localizes CD47 protein to the endoplasmic reticulum [an interior cellular membrane]. CD47 protein localization occurs post-translationally and independently of RNA localization”. The authors propose that the long 3'-UTR acts as a scaffold to recruit various proteins to the site of translation. The interaction of some of these proteins with the newly translated protein results in its localization to the plasma membrane. Importantly, “We also show that CD47 protein has different functions depending on whether it was generated by the short or long 3' UTR isoforms. Thus, alternative polyadenylation contributes to the functional diversity of the proteome without changing the amino acid sequence”. One of the key proteins involved in this localization binds to thousands of mRNAs, so “3' UTR-dependent protein localization has the potential to be a widespread trafficking mechanism for membrane proteins” (Berkovits and Mayr 2015, doi:10.1038/nature14321).
- “Certain tissues preferentially produce mRNAs of a certain length. Brain, pancreatic islet, ear, bone marrow, and uterus showed a preference for distal PASs [polyadenylation sites], leading to longer 3'-UTRs. Retina, placenta, ovary, and blood showed a preference for proximal PASs ... Although most of the transcripts detected in the brain contain distal PASs, the transcripts that are highly abundant generally show a preference for proximal PASs and have short 3'-UTRs. Other studies showed that the choice between a distal and a proximal PAS was modulated during differentiation and development. Progressive lengthening of 3'-UTRs was shown for most of the transcripts during cell differentiation and during embryonic development. By contrast, shortening was observed during proliferation and during reprogramming of somatic cells” (Klerk and ’t Hoen 2015, doi:10.1016/j.tig.2015.01.001).
- “The C/P [cleavage and polyadenylation] machinery is composed of 15–20 core polypeptides, including four protein complexes and several single proteins”. In addition, “a growing number” of RNA-binding proteins [RBPs] have been found to work with the core proteins to regulate cleavage and polyadenylation. Some of these RBPs prevent binding of core proteins, and some recruit such proteins. And, of course, these proteins are subject to regulation in turn: for example, “expression of a substantial fraction of the core factors is highly regulated during embryonic development, reprogramming of differentiated cells, and differentiation of myoblasts” (Tian and Manley 2013).
- “Polyadenylation is not only a fundamental step in mRNA biogenesis but is also highly regulated and networked with other aspects of gene expression. Interestingly, recent evidence indicates that the choice of polyadenylation site in many mRNAs changes in response to cell growth, developmental cues or oncogene activation. The resultant shortening of an mRNA’s 3'-untranslated region can remove a variety of regulatory elements, including miRNA target sites, and thereby dramatically alter gene expression patterns” (Dickson and Wilusz 2010; Mangone, Manoharan, Thierry-Mieg et al. 2010).
- Alternative polyadenylation, by altering the 3'-untranslated region of a transcript, can affect the localization of the transcript within the cytoplasm (which in turn bears on the functional effect of the transcript and the protein produced from it) (Shi 2012).
- Again, the length of the 3'-untranslated region can affect translation efficiency. “mRNAs of the polo gene are alternatively polyadenylated, and the isoform with longer 3' UTR is translated more efficiently. Interestingly, when the distal PAS is genetically disrupted so that only the short APA isoform is produced, the transgenic flies die at the pupa stage due to proliferation defects in the precursor cells of the abdomen (Shi 2012).
- Because polyadenylation sites can be located in exons — and, moreover, in different exons — the result can be the production of distinct proteins. An analysis of the human transcriptome “found that over 5000 human genes produced APA [alternative polyadenylation] isoforms that have differences in their coding regions, and half of these APA events showed tissue-specific profiles. Therefore, like alternative splicing, APA significantly expands the proteome diversity”. The different isoforms may have profoundly different functions, and may also play a role in regulating the amounts of protein produced (Shi 2012).
- Tissue-specific polyadenylation of a gene in the mammalian central nervous system — an imprinted gene expressed paternally — results in extension of the transcript more than 10,000 base pairs downstream into the neighboring gene, which happens to be an antisense gene. This results in the neighboring gene being preferentially expressed from the maternal allele in central nervous system tissues. “Our results propose a new mechanism to regulate allelic usage in the mammalian genome, via tissue-specific alternative polyadenylation and transcriptional interference in sense-antisense pairs at imprinted loci” (MacIsaac, Bogutz, Morrisy and Lefebvre 2012).
- “Modulation of alternative polyadenylation in metazoans is an important means of regulating gene expression. One way that such regulation can be achieved is through altered expression of core components of the 3′ processing machinery. The archetypal example is control of immunoglobulin heavy chain poly(A) site choice by regulating expression of CstF64 [part of a cleavage stimulation factor]...Another recently discovered mechanism works through [splicing factor] U1snRNP inhibition of cleavage at cryptic poly(A) sites, probably by interacting with core cleavage-polyadenylation factors. Our results suggest a related mechanism for alternative polyadenylation regulation; namely, an export adaptor, which interacts with the core 3′ end processing machinery but is not itself a cleavage-polyadenylation factor, can function as a general modulator of poly(A) site choice” (Johnson, Kim, Erickson and Bentley 2011).
- “Our analysis reveals that yeast histone mRNAs have shorter than average PolyA tails and the length of the PolyA tail varies during the cell cycle; S-phase histone mRNAs possess very short PolyA tails while in G1, the tail length is relatively longer...Thus, histone mRNAs are distinct from the general pool of yeast mRNAs and 3'-end processing and polyadenylation contribute to the cell cycle regulation of these transcripts”. Certain 3'-end-processing proteins play a role in this regulation (Beggs, James and Bond 2012).
- “An mRNA 3' processing factor, Fip1, is essential for embryonic stem cell (ESC) self‐renewal and somatic cell reprogramming. Fip1 promotes stem cell maintenance, in part, by activating the ESC‐specific alternative polyadenylation profiles to ensure the optimal expression of a specific set of genes, including critical self‐renewal factors. Fip1 expression and the Fip1‐dependent alternative polyadenylation program change during ESC differentiation and are restored to an ESC‐like state during somatic reprogramming. Mechanistically, we provide evidence that the specificity of Fip1‐mediated alternative polyadenylation regulation depends on multiple factors, including Fip1‐RNA interactions and the distance between alternative polyadenylation sites” (Lackford, Yao, Charles et al. 2014).
- Many proteins are rhythmically expressed in a circadian (24-hour) manner despite the fact that their corresponding mRNAs are produced via gene transcription at a more or less constant rate. This involves, at least in part, the periodic shortening of the polyadenylated tails of the mRNAs, storage of the mRNAs for a period, and then lengthening (re-adenylation) of the tails followed by a new round of translation. (Incidentally: “many steps in the lifetime of an mRNA can be subject to circadian regulation. These steps include regulation of pre-mRNA splicing efficiency, alternative splicing, poly(A) site selection, mRNA editing, nuclear export, mRNA stability, and translation efficiency”.) (Gotic and Schibler 2012)
- It’s not only protein-coding genes that are affected by alternative polyadenylation. Regarding long noncoding RNAs: “Most if not all of these RNAs also use C/P [cleavage and polyadenylation] for 3'-end processing. ... lncRNA genes are more likely than mRNA genes to have alternative pAs [polyadenylation sites] in upstream regions” (thus allowing for radical changes in the length of the long noncoding RNA, and presumably also in its function) (Tian and Manley 2013).
- “Alternative cleavage and polyadenylation (APA) allows genes that contain multiple cleavage and polyadenylation signals (CPAs) to encode multiple RNA isoforms and has an important role in the regulation of gene expression. Now, Neve et al. report the differential regulation of APA isoforms in cytoplasmic and nuclear RNA fractions of human cell lines. APA isoforms with shorter 3ʹ untranslated regions (UTRs), owing to cleavage at promoter-proximal versus promoter-distal CPAs, were over-represented in the cytoplasm in all non-neuronal cell lines analysed, but not in neuroblastoma-derived cells. Further experiments indicated that the nuclear retention of distal CPA isoforms (with longer 3ʹ UTRs) can be partly attributed to incomplete splicing, and demonstrated that the nuclear endoribonuclease DICER1 controls subcellular APA profiles by influencing CPA site selection and through microRNA-mediated stabilization” (Anonymous 2016, doi:10.1038/nrg.2015.33, summarizing work by Neve et al. 2016, doi:10.1101/gr.193995.115).
- “Stress induces an accumulation of genes with differentially expressed polyadenylated mRNA isoforms in human cells. Specifically, stress provokes a global trend in polyadenylation site usage toward decreased utilization of promoter-proximal poly(A) sites in introns or ORFs and increased utilization of promoter-distal polyadenylation sites in intergenic regions. This extensively affects gene expression beyond regulating mRNA abundance by changing mRNA length and by altering the configuration of open reading frames” (Hollerer, Curk, Haase et al. 2016, doi:10.1261/rna.055657.115).
- “Recent observations showed that nascent RNA polymerase II transcripts, pre-mRNAs, and noncoding RNAs are highly susceptible to premature 3′-end cleavage and polyadenylation (PCPA) from numerous intronic cryptic polyadenylation signals (PASs). The importance of this in gene regulation was not previously appreciated as PASs, despite their prevalence, were thought to be active in terminal exons at gene ends. Unexpectedly, antisense oligonucleotide interference with U1 snRNA base-pairing to 5′ splice sites, which is necessary for U1 snRNP’s (U1) function in splicing, caused widespread PCPA in metazoans. This uncovered U1’s PCPA suppression activity, termed telescripting, as crucial for full-length transcription in thousands of vertebrate genes, providing a general role in transcription elongation control. (Venters, Oh, Di et al. 2019, doi:10.1101/cshperspect.a032235)
- “The proper subcellular localization of RNAs and local translational regulation is crucial in highly compartmentalized cells, such as neurons. RNA localization is mediated by specific cis-regulatory elements usually found in mRNA 3' UTRs. Therefore, processes that generate alternative 3' UTRs — alternative splicing and polyadenylation — have the potential to diversify mRNA localization patterns in neurons. ... Our analysis identified 593 genes with differentially localized 3' UTR isoforms. In particular, we have shown that two isoforms of Cdc42 gene with distinct functions in neuronal polarity are differentially localized between neurites and soma of mESC-derived [mouse embryonic stem cell-derived] and mouse primary cortical neurons, at both mRNA and protein level. ... we have identified the role of alternative 3' UTRs and mRNA transport in differential localization of alternative CDC42 protein isoforms. Moreover, we ... identify isoform-specific Cdc42 3' UTR-bound proteome with potential role in Cdc42 localization and translation. Our analysis points to usage of alternative 3' UTR isoforms as a novel mechanism to provide for differential localization of functionally diverse alternative protein isoforms” (Mattioli, Rom, Franke et al. 2019, doi:10.1093/nar/gky1270).
-
Crosstalk: splicing and polyadenylation.
- “Splicing and C/P [cleavage and polyadenylation] are frequently interconnected. This is indicated, for example, by the interactions between key factors involved in these two processes”. Likewise, transcriptional activity (the variable dynamics and structure of the transcribing enzyme, RNA polymerase II) affects cleavage and polyadenylation. And, again, processes bearing on chromatin structure and modification are correlated with cleavage and polyadenylation (Tian and Manley 2013).
-
Crosstalk: deadenylation and decapping.
- Evidence “suggests that the coupling of deadenylation with decapping is, in part, a direct consequence of coordinated assembly of decay factors” (Alhusaini and Coller 2016, doi:10.1261/rna.054742.115).
-
Crosstalk: miRNAs and polyadenylation.
- Alterations in the 3'-untranslated region due to polyadenylation can affect mRNA stability and translation efficiency. For example, many miRNA target sites lie in the 3'-untranslated region — often downstream from the first polyadenylation site. So the choice of polyadenylation site can eliminate miRNA mediated degradation of the transcript. As another example: the sheer length of the 3'-untranslated region can result in nonsense-mediated decay of the mRNA transcript (Shi 2012).
- “We demonstrate that miR-34a represses HDM4, a potent negative regulator of [tumor suppressor] p53, creating a positive feedback loop acting on p53. In a Kras-induced mouse lung cancer model, miR-34a deficiency alone does not exhibit a strong oncogenic effect. However, miR-34a deficiency strongly promotes tumorigenesis when p53 is haploinsufficient, suggesting that the defective p53–miR-34 feedback loop can enhance oncogenesis in a specific context. The importance of the p53/miR-34/HDM4 feedback loop is further confirmed by an inverse correlation between miR-34 and full-length HDM4 in human lung adenocarcinomas. In addition, human lung adenocarcinomas generate an elevated level of a short HDM4 isoform through alternative polyadenylation. This short HDM4 isoform lacks miR-34-binding sites in the 3' untranslated region, thereby evading miR-34 regulation to disable the p53-miR-34 positive feedback. Taken together, our results elucidated the intricate cross-talk between p53 and miR-34 miRNAs and revealed an important tumor suppressor effect generated by this positive feedback loop” (Okada, Lin, Ribeiro et al. 2014).
-
Future expectations. “RBPs [RNA-binding proteins] and core
factors appear to interact with different cis elements around the
pA [site of cleavage and polyadenylation], and pA usage seems to be
determined in a combinatorial manner. How regulation of these
factors, including post-translational modifications, leads to APA
[alternative cleavage and polyadenylation] needs to be explored.
... How other proteins interacting with the C/P [cleavage and
polyadenylation] machinery can change APA needs to be established.
Finally, a clearer picture of APA regulation by chromatin
organization, histone modifications, and DNA methylation, is
expected to emege in the coming years” (Tian and Manley 2013).
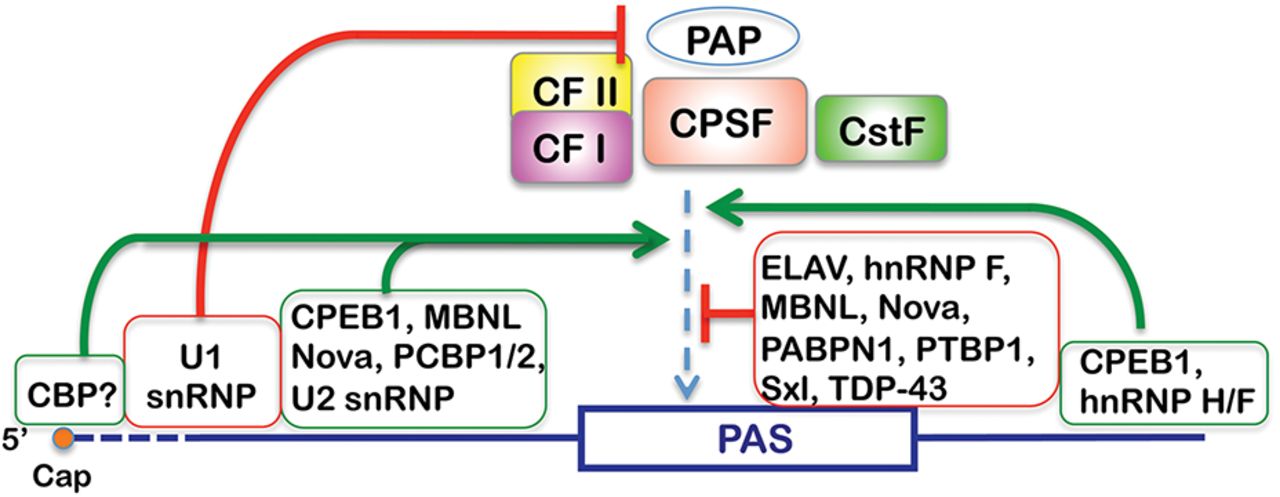
Two years later: “Recent studies suggest that the protein–RNA interaction network involved in PAS [polyadenylation site] recognition is more complex than previously thought, which raises many important questions for future studies”. Caption of figure at right: “Context-dependent regulation of PAS recognition. Regulatory factors bound at different locations relative to the core PAS sequence have different effects on PAS recognition by the mRNA 3′ processing factors. Positive effects are indicated by an arrow, and negative effects are indicated by a vertical line” (Shi and Manley 2015, doi:10.1101/gad.261974.115).
- “Alternative polyadenylation (APA)-associated genetic variants have been proposed to impact diverse human phenotypes and disorders. In a recent study, Li et al. established a landscape of 3′-untranslated region (UTR) APA quantitative trait loci (3'aQTLs) across multiple human tissues, revealing substantial 3'aQTLs that contribute to complex human traits and diseases” (Table of Contents blurb for Fang and Li 2021, doi:10.1016/j.tig.2021.06.002).
-
RNA 3'-end oligouridylation
Oligouridylation is the addition of uridines to the tail end of RNAs,
which can occur in many species, including humans. “Our data revealed
widespread nontemplated [that is, not coded for in DNA] nucleotide
addition to the 3' ends of many classes of RNA, with short stretches of
uridine being the most frequently added” (Choi, Patena, Leavitt and
Mcmanus 2012).
- “The 3' end of U6 snRNA [involved in mRNA splicing] is stabilized after the addition of nontemplated uridines” (Choi, Patena, Leavitt and Mcmanus 2012).
- “The destabilizing effect of uridine addition to mRNAs has been observed in yeast and mammalian cells...After DNA replication is complete, degradation of histone mRNA is initiated by nontemplated oligouridylation to coordinate histone expression with DNA abundance” (Choi, Patena, Leavitt and Mcmanus 2012).
- “Oligouridylation is associated with miRNA biogenesis, function, and turnover” (Choi, Patena, Leavitt and Mcmanus 2012).
- RNA polyuridylation can occur after deadenylation. The short uridine tract draws attention from the exoribonuclease Dis3L2, which proceeds to degrade the RNA from the 3' end. The signifiance of this degradation pathway is indicated by the fact that a mutation in Dis3L2 is “associated with Perlman’s fetal overgrowth syndrome and a propensity for Wilm’s tumour development”. Dis3L2 also aids in maintenance of stem cell pluripotency by inhibiting expression of the let-7 miRNA. It does so by degrading let-7 precursors with uridylate tails (Gallouzi and Wilusz 2013).
- “Uridylation of mRNAs is widespread and conserved among eukaryotes. Uridylation has a fundamental role in mRNA decay and triggers both 5'–3' and 3'–5' degradation. Uridylation can also ‘repair’ mRNA extremities as shown for replication-dependent histone mRNAs during S-phase in humans and for deadenylated mRNAs in Arabidopsis” (Scheer, Zuber, De Almeida and Gagliardi 2016, doi:10.1016/j.tig.2016.08.003).
- “3ʹuridylation of LINE-1 mRNAs by terminal uridyltransferases (TUTases) inhibits LINE-1 retrotransposition” (Strzyz 2018, doi:10.1038/s41580-018-0058-2).
-
Transcript leaders (5'-untranslated regions, or 5'-UTRs)
Transcript leaders, or 5'-untranslated regions (UTRs) are (in part)
nucleotide sequences attached to the 5' end of a premature mRNA
after its transcription, to which a methyl-7-guanosine (m7G)
cap is added. Preparatory to translation, a cap-binding protein
complex binds to the cap and facilitates formation of a pre-translation
initiation complex (including a small ribosomal subunit). This complex
then “scans in a net 5'-to-3' direction until it locates a start codon
(AUG), triggering complex rearrangements that eventually result in
formation of an elongating 80S ribosome”, beginning the process of
actual translation, yielding a protein (Arribere and Gilbert 2013).
Recent studies “have revealed widespread post-transcriptional
regulation by TLs [transcript leaders]”. “Transcript leaders can have
profound effects on mRNA translation and stability”. “We identified
[in yeast] hundreds of cases where one gene encodes multiple TL
isoforms, and showed that the majority of these variants are associated
with distinct translational activities in vivo”. TL diversity in
mammals is “quite common” compared to the relatively low levels of
diversity in yeast (Arribere and Gilbert 2013).
“RNA caps are deposited at the 5′ end of RNA polymerase II transcripts.
This modification regulates several steps of gene expression, in addition
to marking transcripts as self to enable the innate immune system to
distinguish them from uncapped foreign RNAs, including those derived from
viruses. Specialized immune sensors, such as RIG-I and IFITs, trigger
antiviral responses upon recognition of uncapped cytoplasmic transcripts.
Interestingly, uncapped transcripts can also be produced by mammalian
hosts. For instance, 5′-triphosphate RNAs are generated by RNA polymerase
III transcription, including tRNAs, Alu RNAs, or vault RNAs. These RNAs
have emerged as key players of innate immunity, as they can be recognized
by the antiviral sensors. Mechanisms that regulate the presence of
5′-triphosphates, such as 5′-end dephosphorylation or RNA editing,
prevent immune recognition of endogenous RNAs and excessive inflammation”
(Avila-Bonilla and Macias 2024, doi:10.1261/rna.079942.124).
- Evidence suggests that there is widepsread heterogeneity in TLs, and therefore also considerable regulation potential. In yeast, “more than 99% of genes analyzed by Miura and 95% of genes in Zhang and Dietrich had more than one TL” (Arribere and Gilbert 2013).
- Some translation leaders have start codons in them, which leads to decreased translation initiation from the main start codon in the protein-coding region of the gene, and to nonsense-mediated mRNA decay (NMD) of the product of translation. Some of these “upstream” start codons “have well-characterized translational regulatory functions, including those found in the TLs of the stress-responsive transcription factors GCN4 and ATF4” (Arribere and Gilbert 2013).
- Very short TLs, which were observed in hundreds of yeast genes, “lead to [translation] initiation at downstream AUGs, often culminating in nonsense-mediated mRNA decay.
- “Other TLs allow specific genes to be efficiently translated under conditions of widespread translational inhibition” (Arribere and Gilbert 2013).
- In humans “the majority of TL variants showed tissue-specific expression patterns. Importantly, because most intragenic TL variants do not change the coding potential of the mRNA, their influences must be felt during the post-transcriptional life of the mRNA, namely, during translation, [mRNA] localization, and/or decay” (Arribere and Gilbert 2013).
-
RNA cleavage
This is different from the mRNA cleavage or degradation usually spoken
of in connection with microRNAs and siRNAs. It turns out that many
RNAs are cleaved, not just for down-regulation, but in order to achieve
a wide range of different RNAs, with distinct functions in the organism.
- “Post-transcriptional cleavage events are widespread, conserved among eukaryotes, and generate a range of small RNAs and long coding and noncoding RNA (ncRNA) transcripts...the secondary capping of cleaved transcripts is a regulated process that is conserved between species and regulated in a developmental-stage and tissue-specific manner. ... The cleavage pathway has significant impact in remodeling the transcriptome. We conclude that post-transcriptional RNA cleavage is a common mechanism that, alongside transcription initiation, termination, alternative splicing, and editing, plays a significant part in the diversification of both the coding and noncoding transcriptional repertoire of the genome” (Mercer, Dinger, Bracken et al. 2010). How the cleavage occurs isn’t yet well-established, although microRNAs may well be involved.
- “RNA fragmentation significantly expands the already extraordinary spectrum of transcripts present within eukaryotic cells, and also calls into question how the ‘gene’ should be defined” (Tuck and Tollervey 2011).
-
RNA splicing
-
Nuclear export and RNA localization
During the process of transcription and afterward, a messenger RNA comes
into association with numerous proteins, forming a dynamic messenger
ribonucleoprotein complex that goes through continual transformations in
order to achieve various functions on the way from transcription through
editing and splicing, to export from the nucleus and translation.
(Translation of the mRNAs occurs in the cytoplasm.)
- mRNA localization is mentioned as part of many topics under POST-TRANSCRIPTIONAL DECISION-MAKING above. For example, alternative splicing and polyadenylation can affect mRNA localization.
- What a protein “means” in the organism — and therefore what a protein is — depends on its function, and while such things as alternative RNA splicing and post-translational modifications can affect a protein’s function, so, too, can the locale to which the protein is directed in order to be translated: “Although many proteins are localized after translation, asymmetric protein distribution is also achieved by translation after mRNA localization. Why are certain mRNA transported to a distal location and translated on-site? ... Our findings suggest that asymmetric protein distribution by mRNA localization enhances interaction fidelity and signaling sensitivity. Proteins synthesized at distal locations frequently contain intrinsically disordered segments. These regions are generally rich in assembly-promoting modules and are often regulated by post-translational modifications. Such proteins are tightly regulated but display distinct temporal dynamics upon stimulation with growth factors. Thus, proteins synthesized on-site may rapidly alter proteome composition and act as dynamically regulated scaffolds to promote the formation of reversible cellular assemblies. Our observations are consistent across multiple mammalian species, cell types and developmental stages, suggesting that localized translation is a recurring feature of cell signaling and regulation” (Weatheritt, Gibson and Babu 2014).
- “An increasing number of studies indicate that PTMs contribute to the coupling and coordination of mRNA export steps by regulating the dynamic association of proteins with maturing mRNPs [messenger RNA-protein complexes]. Indeed, from transcription to export, proteins signal their transition from one stage to the next through PTMs [post-translational modifications] that inhibit or trigger interactions with sequential partners”. These modifications add “a new level of regulation to the process of gene expression” (Tutucci and Stutz 2011).
- “Mature mRNAs have been thought to reside predominantly in the cytoplasm, where they serve as templates for protein translation. Bahar Halpern et al. analysed cytoplasmic versus nuclear mRNA pools in pancreas and liver cells and found that, in fact, fully mature mRNAs of a significant fraction of genes (including various metabolic genes) are found in higher amounts in the nucleus than in the cytoplasm. This was attributed to low mRNA export rates in comparison to cytoplasmic degradation rates. Computer modelling based on these data indicated that such a nuclear accumulation of mRNAs might dampen gene expression noise, which originates from the pulsatile nature of transcription. Thus, mRNA nuclear retention could confer robustness to the process of gene expression, without the need to alter the steady-state levels of mRNA” (Strzyz 2016, doi:10.1038/nrm.2016.4).
-
RNA-protein complexes (RNPs)
“Ultimately, the fate of any given mRNA is determined by the ensemble of all
associated RNA-binding proteins (RBPs), non-coding RNAs and metabolites
collectively known as the messenger ribonucleoprotein particle (mRNP) ...
The mRNA-bound proteome is more complex than previously anticipated and
comprises up to 1000 RBPs. Because there are many mRNA-interacting factors,
and each mRNA is the blueprint of a particular protein, the resulting
ribonucleoprotein particles (mRNPs) are likely to be unique in their
composition. The ‘mRNP code’ concept implies that specific sets of
proteins, non-coding RNAs, and other molecules bind to individual mRNAs and
control their fate and function in every cell. The mRNP code is highly
dynamic and reflects the functional status of each mRNA. Previously unknown
RNA-binding domains show unconventional modes of RNA-protein interactions”
(Gehring, Wahle and Fischer 2017, doi:10.1016/j.tibs.2017.02.004).
Regarding the almost unlimited complexity and regulatory potential of
RNA-protein interaction, which seems to illustrate the general biochemical
principle that “almost anything can do at least something with almost
anything”:
“Prediction of (m)RBPs solely from their amino acid sequence or the presence
of known RNA-binding domains has proven difficult or misleading. On the one
hand, not all designated RNA-binding domains indeed mediate the interaction
with RNA; some contact other proteins instead. On the other, recently
established catalogs of RBPs include many factors that have not been
previously linked to RNA. Furthermore, up to one third of candidate RBPs do
not contain ‘classical’ RNA-binding domains. In fact, domains previously
implicated in scaffolding functions now turn out to confer specific RNA
binding. For example, the NHL domain of the Drosophila protein BRAT was
recently shown to form an unconventional RNA-binding module that binds to
single-stranded RNA in a sequence-specific manner via a positively charged
platform. Likewise, a WD40 repeat, which typically forms a propeller-like
protein interaction scaffold in signaling molecules, has recently been shown
to bind to RNA in a specific manner in the context of the protein Gemin5.
Even unstructured protein regions such as arginine-glycine-glycine (RGG)
stretches can mediate RNA binding. It is therefore very difficult to
predict whether a protein is indeed an RNA binder without experimental
validation”
(Gehring, Wahle and Fischer 2017, doi:10.1016/j.tibs.2017.02.004).
“Many RBPs such as members of the heterogeneous nuclear (hn)RNP protein
family contain more than one RNA-binding domain or even combinations of
several different types thereof. This enables binding to longer stretches of
RNA and typically leads to an increased affinity and specificity of the RBP.
Alternatively, multiple RNA-binding domains may also be combined to
recognize non-contiguous binding sites on mRNA, thereby assisting
topological organization of mRNAs and/or properly positioning other
components of the mRNP. Indeed, recent studies suggest that the
combinatorial use of RNA binding domains in proteins may be even more common
than was previously assumed”
(Gehring, Wahle and Fischer 2017, doi:10.1016/j.tibs.2017.02.004).
“the mRNP may also interact with small organic molecules or ions. The
enzymatic cofactor thiamin pyrophosphate, for example, binds to structured
RNA elements (riboswitches) within introns of specific mRNAs and regulates
their processing. Ions, by contrast, often contribute to the stabilization
of RNA secondary and tertiary structure, or regulate the binding of proteins
to their mRNA target, as described for zinc-finger proteins”
(Gehring, Wahle and Fischer 2017, doi:10.1016/j.tibs.2017.02.004).
RNA-protein complexes are mentioned throughout this document, and represent
an entire universe of gene-regulatory functions, only a small percentage of
which are alluded to anywhere here. In particular, a number of small
nuclear ribonucleoproteins (snRNPs) play decisive roles in RNA splicing.
See the extensive notes under Alternative
splicing. Also, proteins interacting with RNAs can play roles in
deadenylation, readenylation, uridylation, editing, and/or base
modifications, topics covered elsewhere in this document.
In sum, “the emerging picture is that the mRNP can remain stable during
particular periods of gene expression but will be remodeled when the mRNA
enters the next functional stage of its life cycle. Similar to the initial
establishment of the mRNP, its remodeling is an important process during the
life of an mRNA, which may involve trans-acting factors and complex
molecular mechanisms. This remodeling can occur in an active, ATP-driven
manner, as exemplified by the action of helicases, or in a passive manner by
simple association and dissociation events”
(Gehring, Wahle and Fischer 2017, doi:10.1016/j.tibs.2017.02.004).
“mRNPs always represent an ensemble of many different factors that may act
in a combinatorial manner and whose functions need to be coordinated. How
this is achieved is currently only beginning to be understood and is
certainly a major challenge for future studies”
(Gehring, Wahle and Fischer 2017, doi:10.1016/j.tibs.2017.02.004).
-
Small nuclear ribonucleoproteins (snRNPs)
These form part of the spliceosome complex (see
“RNA splicing” and
“Alternative splicing” above), but they
are now being found to have functions separate from splicing.
- The U1 small nuclear ribonucleoprotein protects pre-mRNAs from premature cleavage and polyadenylation (Kaida, Berg, Younis et al. (2010).
- The U2 snRNP plays a role in the 3'-end formation of histone mRNAs.
- (See under “Alternative cleavage, polyadenylation, and deadenylation” above.)
-
Exon junction complexes
The multiprotein “exon junction complex” (EJC) is deposited by spliceosomes
onto mRNAs following splicing. It is located at a conserved position 24
nucleotides upstream of the spliced junction, “and adopts a unique
structure, which can both stably bind to mRNAs and function as an anchor for
diverse processing factors. Recent findings revealed that in addition to its
established roles in nonsense-mediated mRNA decay, the EJC is involved in
mRNA splicing, transport and translation. While structural studies have shed
light on EJC assembly, transcriptome-wide analyses revealed differential EJC
loading at spliced junctions. Thus, the EJC functions as a node of
post-transcriptional gene expression networks, the importance of which is
being revealed by the discovery of increasing numbers of EJC-related
disorders” (Hir, Saulière and Wang 2016, doi:10.1038/nrm.2015.7).
-
RNA-binding proteins and RNA helicases
Most information relative to these proteins is contained under specific
functional headings, such as “Nuclear export”,
and “Alternative splicing”.
- A large number of RNA-binding proteins play diverse and crucial roles in coordinating several levels of mRNA regulation on the way from transcription to translation. These include splicing, transport, stability, proper localization, and translation itself (Keene 2007). We could have no functioning products of gene expression without the nuanced performances of these proteins.
- RNA helicases are enzymes that bind to and remodel RNA or RNA-protein complexes. They use energy from ATP to unwind RNA duplexes, displace other regulatory proteins from RNA, act as chaperones to ensure proper folding of RNA molecules, and engage in the proofreading of RNA during the splicing reaction. They typically work in the context of complex molecular assemblies, interacting with many other proteins. The functional consequences of this activity are as yet little understood (Jankowsky 2011).
- Sex-specific RNA binding by proteins. In Drosophila many genes appear to be regulated in a way that depends on sex-specific differences in the untranslated regions (UTRs) of mRNA. Untranslated regions can vary as a result, for example, of alternative polyadenylation of 3'-UTR, and the use of alternative promoters in transcription, which changes the 5'-UTR. A Drosophila protein, UNR, binds to many RNA UTRs based on sex-specific differences in UTRs, thereby regulating gene expression in a sex-specific manner (Mihailovich, Wurth, Zambelli et al. 2011).
- “Two new studies show that RNA-binding proteins can mediate distinct and beneficial effects to cells by binding to the extensive double-stranded RNA (dsRNA) structures of inverted-repeat Alu elements (IRAlus). One study reports stress-induced export of the 110-kDa isoform of the adenosine deaminase acting on RNA 1 protein (ADAR1p110) to the cytoplasm, where it binds IRAlus so as to protect many mRNAs encoding anti-apoptotic proteins from degradation. The other study demonstrates that binding of the nuclear helicase DHX9 to IRAlus embedded within RNAs minimizes defects in RNA processing”. “This 'yin and yang' of IRAlus is the result of competition between dsRNA-binding proteins that stabilize IRAlus and mediate IRAlu functions and other dsRNA-binding proteins and helicase enzymes that destabilize IRAlus and inhibit these functions” (Elbarbary and Maquat 2017, doi:10.1038/nsmb.3416).
- “Quaking protein isoforms arise from a single Quaking gene and bind the same RNA motif to regulate splicing, translation, decay, and localization of a large set of RNAs. However, the mechanisms by which Quaking expression is controlled to ensure that appropriate amounts of each isoform are available for such disparate gene expression processes are unknown. Here we explore how levels of two isoforms, nuclear Quaking-5 (Qk5) and cytoplasmic Qk6, are regulated in mouse myoblasts. We found that Qk5 and Qk6 proteins have distinct functions in splicing and translation, respectively, enforced through differential subcellular localization. We show that Qk5 and Qk6 regulate distinct target mRNAs in the cell and act in distinct ways on their own and each other's transcripts to create a network of autoregulatory and cross-regulatory feedback controls. Morpholino-mediated inhibition of Qk translation confirms that Qk5 controls Qk RNA levels by promoting accumulation and alternative splicing of Qk RNA, whereas Qk6 promotes its own translation while repressing Qk5. This Qk isoform cross-regulatory network responds to additional cell type and developmental controls to generate a spectrum of Qk5/Qk6 ratios, where they likely contribute to the wide range of functions of Quaking in development and cancer.” (Fagg, Liu, Fair et al. 2017, doi:10.1101/gad.302059.117)
-
mRNA coordinators
“mRNA coordinators” is the proposed name for a class of proteins that
apparently mediate crosstalk between transcription and translation.
- In yeast the proteins Rpb4p and Rpb7p form a heterodimer (Rpb4/7) that moves between the nucleus and the cytoplasm. Its association with the transcribing enzyme, RNA Polymerase II, in the nucleus leads to its involvement in transcription initiation, elongation, and polyadenylation. At some point it interacts directly with the mRNA transcript. Following transcription, Rpb4/7 is exported to the cytoplasm, where it stimulates translation by interacting with a translation initiation factor.
- Rpb4/7 also stimulates shortening of the polyadenylated tail of the mRNA, an action that leads to mRNA degradation
- “We propose that Rpb4/7, through its interactions at each step in the mRNA life-cycle, represents a class of factors, ‘mRNA coordinators’, which integrate the various stages of gene expression into a system” (Harel-Sharvit, Eldad, Haimovich et al. 2010).
-
mRNA -> mRNA regulation
- "regulatory elements within mRNAs can act in trans to influence the behavior of other mRNA molecules" (article entitled "trans Regulation: Do mRNAs Have a Herd Mentality?” Wilusz and Wilusz 2010). The means for achieving this are not yet known.
-
Competing endogenous RNAs
Various RNAs can “compete” with each other with their recognition sites for
regulatory molecules such as miRNAs. By this means, for example, the
increase of one kind of RNA — which provides ample recognition sites for an
miRNA that also targets a second RNA — can upregulate the second RNA by
“soaking up” the pool of relevant miRNAs. Given the entire collection of
RNAs in a cell, one can easily imagine an unfathomably complex set of
mutual regulatory interactions going on.
- (To do: Consolidate material from Salmena and others in this section.)
- The method of regulation between a gene and a corresponding pseudogene (see “Pseudogenes” below), presumably works between “any two co-expressed genes...that are regulated by the same non-coding RNA” such as miRNAs. The one gene’s mRNA can act as a “decoy” for miRNAs that might otherwise target the other gene’s mRNA (Poliseno, Salmena, Zhang et al. 2010).
- The same sort of regulation, it turns out, can be effected between some long noncoding RNAs (see “Long noncoding RNAs”) and pseudogenes (see “Pseudogenes”).
- The large intergenic noncoding RNA, linc-RoR “maintains human embryonic stem cell self-renewal by functioning as a sponge to trap [the microRNA,] miR-145, thus regulating core pluripotency factors Oct4, Nanog, and Sox2” (doi:10.1016/j.devcel.2013.03.020). In particular: “The embryonic stem cell transcriptional and epigenetic networks are controlled by a multilayer regulatory circuitry, including core transcription factors, posttranscriptional modifier microRNAs (miRNAs), and some other regulators. ... Here, we demonstrate that a lincRNA [large intergenic noncoding RNA], linc-RoR, may function as a key competing endogenous RNA to link the network of miRNAs and core transcription factors, e.g., Oct4, Sox2, and Nanog. We show that linc-RoR shares miRNA-response elements with these core transcription factors and that linc-RoR prevents these core transcription factors from miRNA-mediated suppression in self-renewing human embryonic stem cells” (doi:10.1016/j.devcel.2013.03.002).
- To demonstrate the reality of competing endogenous RNA networks, artificial miRNA sponges have been introduced into cells, both in vitro and in vivo, and have proven effective in de-repressing miRNA targets. “Intriguingly, although sponges with perfectly complementary miRNA-binding sites have been shown to be effective, ‘bulged sponges’, which include a central bulge and hence bind miRNAs with imperfect complementarity, have been demonstrated to sequester miRNAs with greater efficacy. This may be partly due to the fact that, unlike perfectly complementary targets, imperfect targets are not immediately degraded and are thus able to reduce miRNA bioavailability until the mRNA is destabilized by other factors” (Tay, Rinn and Pandolfi 2014).
- Looking at data on transcriptome-wide changes following knockdown of 100 long noncoding RNAs in mouse embryonic stem cells, one research group wondered how much of the transcript-level change was related to the loss of the long noncoding RNAs as competitors with mRNAs for binding by miRNAs (which are particularly abundant in stem cells). Upon depleting mouse stem cells of miRNAs, they found that more than 50% of the long noncoding RNAs and the mRNAs with which the noncoding RNAs shared miRNA target sequences were up-regulated coordinately, thus demonstrating the role of miRNAs in the transcriptional change. The “miRNA-dependent mRNA targets of each lncRNA tended to share common biological functions. Post-transcriptional miRNA-mediated crosstalk between lncRNAs and mRNA, in mESCs, is thus surprisingly prevalent, conserved in mammals, and likely to contribute to critical developmental processes” (Tan, Sirey, Honti et al. 2015, doi:10.1101/gr.181974.114).
- “Recent studies in both solid tumors and hematopoietic malignancies showed that ceRNAs have significant roles in cancer pathogenesis by altering the expression of key tumorigenic or tumor-suppressive genes” (Wang, Hou, He et al. 2016, doi:10.1016/j.tig.2016.02.001).
- “We identified a large number of genetic variants that are associated with ceRNA's function ... We call these loci competing endogenous RNA expression quantitative trait loci or ‘cerQTL’ ... We identified many cerQTLs that have undergone recent positive selection in different human populations, and showed that single nucleotide polymorphisms in gene 3'UTRs at the miRNA seed binding regions can simultaneously regulate gene expression changes in both cis and trans by the ceRNA mechanism. We also discovered that cerQTLs are significantly enriched in traits/diseases associated variants reported from genome-wide association studies in the miRNA binding sites, suggesting that disease susceptibilities could be attributed to ceRNA regulation. Further in vitro functional experiments demonstrated that a cerQTL rs11540855 can regulate ceRNA function. These results provide a comprehensive catalog of functional non-coding regulatory variants that may be responsible for ceRNA crosstalk at the post-transcriptional level” (Li, Zhang, Liang et al. 2017, doi:10.1093/nar/gkx331).
- “Widespread mRNA 3′ UTR shortening through alternative polyadenylation promotes tumor growth in vivo. A prevailing hypothesis is that it induces proto-oncogene expression in cis through escaping microRNA-mediated repression. Here we report a surprising enrichment of 3′UTR shortening among transcripts that are predicted to act as competing-endogenous RNAs (ceRNAs) for tumor-suppressor genes. Our model-based analysis of the trans effect of 3′ UTR shortening (MAT3UTR) reveals a significant role in altering ceRNA expression. MAT3UTR predicts many trans-targets of 3′ UTR shortening, including PTEN, a crucial tumor-suppressor gene3 involved in ceRNA crosstalk4 with nine 3′UTR-shortening genes, including EPS15 and NFIA. Knockdown of NUDT21, a master 3′ UTR-shortening regulator2, represses tumor-suppressor genes such as PHF6 and LARP1 in trans in a miRNA-dependent manner. Together, the results of our analysis suggest a major role of 3′ UTR shortening in repressing tumor-suppressor genes in trans by disrupting ceRNA crosstalk, rather than inducing proto-oncogenes in cis. (Park, Ji, Kim et al. 2018, doi:10.1038/s41588-018-0118-8).
- “Global 3'US [3' untranslated region shortening, which can result from alternative polyadenylation] promotes tumour growth in vivo, which was suggested to result from increased stability of oncogene transcripts. Park, Ji et al. now show that 3'US can in fact contribute to the destabilization of tumour suppressors in trans by modulating networks of competing endogenous RNAs (ceRNAs) ... Analysis of 97 breast cancer samples revealed that their global ceRNA network was much smaller — comprising ten times fewer ceRNA pairs — than in control samples, and this reduction was strongly associated with extensive 3′US of ceRNAs. Notably, the extent of 3'US of ceRNAs in tumours was negatively correlated with the expression levels of their partner genes” (Strzyz 2018, doi:10.1038/s41580-018-0032-z).
-
Proteins that bind both DNA and RNA
There is “evidence that a subset of ZF [zinc-finger] proteins live double
lives, binding to both DNA and RNA targets and frequenting both the
cytoplasm and the nucleus. This duality can create an important
additional level of gene regulation that serves to connect transcriptional
and post-transcriptional control”. “Evolution has favored the emergence of
highly complex and interconnected systems to control gene expression”
(Burdach, O’Connell, Mackay and Crossley 2012).
“Tens of thousands of human lncRNAs [long nonprotein-coding RNAs] have been
catalogued, and it is likely that many of them have yet-undiscovered
functions requiring binding to proteins that are currently considered as
DNA-specific binding proteins” (Hudson and Ortlund 2014).
- “DNA- and RNA-binding proteins can bind DNA and RNA simultaneously, allowing the RNA to function as a scaffold to recruit other proteins to a specific DNA locus” (Hudson and Ortlund 2014).
- As DNA-binders, these proteins can act directly as transcription factors. But when binding to RNA, they are sequestered in the cytoplasm and their transcription factor activity is inhibited. So the presence of the relevant RNAs serves to modulate transcription of the protein-regulated genes.
- “Transcription factors other than ZF proteins can also possess dual-binding domains. The protein bicoid has been shown to bind both DNA and RNA through its homeodomain motif, and there are several examples of other transcription factors binding to RNA, albeit through different domains from those responsible for DNA binding. It is also logical to consider that multifunctional DNA/RNA-binding domains could have many and varied roles beyond transcription. Proteins involved in RNA binding, splicing, RNA editing/processing, DNA repair and other nucleic acid binding events could also potentially have dual DNA/RNA-binding functions. The activity of these proteins might be similarly regulated by the presence of alternate DNA or RNA ligands” (Burdach, O’Connell, Mackay and Crossley 2012).
- “RNA can compete with DNA for binding to DNA- and RNA-binding proteins, typically at the same protein interface. In the case of transcription factors, this can reduce promoter occupancy and the transcription of target genes” (Hudson and Ortlund 2014).
- “DNA- and RNA-binding proteins can regulate gene expression at multiple levels. In addition to binding to the promoters of genes to regulate their transcription, DRBPs [DNA- and RNA-binding proteins] can also affect microRNA (miRNA) processing, as well as mRNA stability and translation” (Hudson and Ortlund 2014).
- “RNA interactions by these multifunctional [protein] regulators can also lead to the coupling of transcription and translation through, for example, direct regulation of the translation of the bound mRNA” (Burdach, O’Connell, Mackay and Crossley 2012). This could be just as well listed under DECISION-MAKING RELATING TO TRANSLATION below.
- A more recent review (2014) explains how RNA-binding proteins (RBPs) must be thought of as engaged in multiple, complexly interrelated activities, and certainly cannot be thought of only as RNA regulators. The relevant studies “show that RBPs prevent harmful RNA/DNA hybrids and are involved in the DNA damage response, from DNA repair to cell survival decisions. Indeed, specific RBPs allow the selective regulation of DNA damage response genes at multiple post-transcriptional levels (from pre-mRNA splicing/polyadenylation to mRNA stability/translation) and are directly involved in DNA repair. These multiple activities are mediated by RBP binding to mRNAs, nascent transcripts, noncoding RNAs, and damaged DNA”. And again: “We propose that DNA damage-induced relocalization of multifunctional RBPs allows the coordinated regulation of various aspects of RNA and DNA metabolism (e.g., DNA repair and DNA damage response gene expression” (Dutertre, Lambert, Carreira et al. 2014).
- “Pumilio proteins bind an extensive network of mRNAs and repress protein expression by inhibiting translation and promoting mRNA decay. Opposingly, in certain contexts, they can activate protein expression. Pumilio proteins also regulate noncoding (nc)RNAs. The ncRNA, ncRNA activated by DNA damage ( NORAD), can in turn modulate Pumilio activity. Genetic analysis provides new insights into Pumilio protein function. They are essential for growth and development. They control diverse processes, including stem cell fate, and neurological functions, such as behavior and memory formation. Novel findings show that their dysfunction contributes to neurodegeneration, epilepsy, movement disorders, intellectual disability, infertility, and cancer” (Goldstrohm, Hall and McKenney 2018, doi:10.1016/j.tig.2018.09.006).
-
RNA granules
- Stress granules (SGs), constituting one class of granule, “regulate mRNA translation and decay”. They seem to be “triage centers that sort, remodel, and export specific mRNA transcripts for reinitiation, decay, or storage. At the same time, SGs contain components with no obvious link to RNA metabolism...and may link SGs to apoptosis” (Anderson and Kedersha 2006, p. 804).
-
Pseudogenes
A pseudogene is usually related to a normal gene (perhaps via duplication),
but — according to a perhaps rather too old definition — has one or more
mutations, or a loss of associated regulatory DNA, preventing either its
transcription or its translation into a protein. Pseudogenes were long
thought to be nonfuctional. However, “Recent advances have established
that the DNA of a pseudogene, the RNA transcribed from a pseudogene, or the
protein translated from a pseudogene can have multiple, diverse functions
and that these functions can affect not only their parental genes but also
unrelated genes. Therefore, pseudogenes have emerged as a previously
unappreciated class of sophisticated modulators of gene expression, with a
multifaceted involvement in the pathogenesis of human cancer” (Poliseno
2012).
Estimates for the number of pseudogenes in the mammalian or human genome
vary considerably, running from 10,000 – 20,000 (at least one for every two
human protein-coding genes).
- It’s been found that the mRNA expressed from a pseudogene can upregulate the translation of the normal gene’s mRNA, by acting as an miRNA “sponge” — that is, by providing “decoy” targets for miRNAs that otherwise would target the normal mRNA for degradation. “The greater the number of pseudogenes that a protein-coding gene has, the more it is protected from miRNAs”. Some cases of such regulation figure in cancer prevention (or, in the case of mutations to the pseudogene, cancer causation). Further, regulation can occur in the opposite direction: the normal mRNA can influence the amount of the pseudogene’s mRNA. (Poliseno, Salmena, Zhang et al. 2010).
- “It had been reported previously that the PTEN pseudogene functions as a miRNA ‘sponge’, similar to the CEBPA lncRNA that acts to sponge DNMT1 away from the CEBPA promoter. Studies to interrogate the PTEN pseudogene in greater detailed determined that this pseudogene also expressed an antisense lncRNA in trans which functions to direct transcriptional gene silencing to the PTEN promoter and control PTEN expression epigenetically. Mechanistically, the PTEN pseudogene expressed antisense lncRNA modulated PTEN transcription by recruiting DNMT3a and EZH2 to the PTEN promoter” (Weinberg and Morris 2016, doi:10.1093/nar/gkw139).
- “Although transcribed pseudogenes may be expressed at much lower levels than their cognate genes, this is counterbalanced by their high degree of shared sequence homology, which results in the conservation of multiple miRNA-binding sites and allows them to compete for the binding of many shared miRNAs simultaneously. Furthermore, it has been suggested that RNA transcripts that contain premature stop codons, such as pseudogenes, may be subjected to nonsense-mediated mRNA decay. This rapid turnover may conceivably lead to their low abundance, as well as the increased degradation of bound miRNAs, enhancing their effectiveness as miRNA sponges” (Tay, Rinn and Pandolfi 2014). See Competing endogenous RNAs above.
- In mice, pseudogenes have been reported “to generate endogenous siRNAs that downregulate the expression of cognate genes through conventional RNA interference” (Poliseno, Salmena, Zhang et al. 2010, citing work by Okamura, Chung and Lai). Pseudogene-derived siRNAs have also been found in protists and plants. They help regulate metabolism and many other functions.
- “the relationship between pseudogenes and their parental counterparts is extremely varied. The pseudogene can promote or inhibit the expression of or enhance or impair the function of the parental gene. It is conceivable that the function of the parental gene is impaired by the pseudogene at one level (for instance, the pseudogenic protein is an allosteric inhibitor of the protein encoded by the parental gene) but is promoted at another level (for instance, the pseudogenic RNA competes with the parental RNA for a microRNA). Pseudogenes could thereby act as sophisticated regulators of their parental counterparts, finely tuning every step of the parental genes’ expression as well as their activity” (Poliseno 2012).
- “Consistent with the notion that they exert biological functions, the expression of pseudogenes is a regulated process. Pseudogene transcripts can be subjected to alternative splicing and have 3' untranslated regions (UTRs) of variable length due to the existence of multiple polyadenylation signals...the global expression profile of pseudogenes differs in different lineages and under different conditions. For example, the pseudogene transcriptome can vary during physiological processes, such as neural differentiation, as well as in association with pathophysiological conditions, such as asthma or HIV infection. Furthermore, various pseudogenes show a spatiotemporal expression pattern distinct from that of their coding counterparts” (Poliseno 2012).
- Researchers who triggered the expression of NF-κB (a transcription factor associated with the inflammatory response) in cultured mouse fibroblasts found that the levels of hundreds of long noncoding RNAs were driven up or down — and 54 of these derived from pseudogenes. As the leader of the research team, Howard Chang, put it, “When a cell is subjected to an inflammatory stress signal, it’s like Night of the Living Dead”. Moreover, different signaling molecules activated different pseudogenes. “They’re not really dead, after all. They just need very specific signals to set them in motion”. Inflammation, if sustained too long, damages healthy tissue. One of the pseudogenes (called “Lethe”) activated during these experiments subsequently served to downregulate NF-κB, thereby reducing the inflammatory response (Papicavoli, Qu, Zhang et al. 2013; Goldman 2013).
-
Drosha-mediated mRNA cleavage
- Drosha is an enzyme involved in the creation of miRNAs. However, it has recently been discovered to act directly in its own right in cleaving mRNA molecules and therefore in regulating protein expression from genes. “Drosha-mediated mRNA cleavage ... adds to an ever-increasing variety of post-transcriptional mechanisms of gene regulation. Such a variety of mechanisms highlights the importance of fine-tuning the expression of genes, rather than simply turning genes on or off” (Chong, Zhang, Cheloufi et al. 2010).
-
RNA degradation
“mRNA degradation is a key controlling factor in gene expression
regulation, possibly as important as transcription factor-mediated
induction of mRNA synthesis” (’t Hoen, Hirsch, de Meijer et al. 2010).
This is true if only because effective, or net, gene expression is in part
a matter of the balance between transcription and mRNA degradation. But
there is much more than that. For example, degradation itself contributes
its own byproducts to further gene regulation: “Recently, it has become
increasingly clear that the composition of the cellular RNA degradome can
be modulated by numerous endogenous and exogenous factors (e.g. by stress).
In addition, instead of being hydrolyzed to single nucleotides, some
intermediates of RNA degradation can accumulate and function as signalling
molecules or participate in mechanisms that control gene expression. Thus,
RNA degradation appears to be not only a process that contributes to the
maintenance of cellular homeostasis but also an underestimated source of
regulatory molecules” (Jackowiak, Nowacka, Strozycki and Figlerowicz 2011).
(There is also the matter of protein degradation; see
“Protein homeostasis network”
below.)
“In recent years, three seemingly distinct aspects of RNA biology — mRNA
N6-methyladenosine modification, alternative 3' end processing and
polyadenylation, and mRNA codon usage — have been linked to mRNA turnover,
and all three aspects function to regulate global mRNA stability in
cis”
• “The 3′ UTRs of many protein-coding genes harbor multiple polyadenylation signals that are differentially selected based on the physiological state of cells, resulting in alternative mRNA isoforms with differing mRNA stability.
• “m6A is the most abundant base modification in eukaryotic mRNA but many functional impacts of m6A on mRNA fate, mRNA stability in particular, have been discovered only recently.
• “Codon usage in mRNA open-reading frames (ORFs) influences gene expression, with the proportion of optimal and nonoptimal codons helping to fine-tune mRNA stability in a process that is coupled to translation” (Chen and Shyu 2016, doi:10.1016/j.tibs.2016.08.014).
Sequence elements in mRNAs “are operated by RNA-binding proteins (RBPs)
and/or miRNA-containing complexes. Based on the large number of RBPs and
miRNAs encoded in metazoan genomes, their complex developmental expression
and that specific RBP and miRNA interactions with mRNAs can lead to
distinct degradation rates, I propose that developmental gene expression is
shaped by a complex ‘mRNA degradation code’ with high information capacity.
Localised cellular events involving the modification of RBP and/or miRNA
target sequences in mRNAs by alternative polyadenylation added to the
activation of specific RBP and miRNA activities via cell signalling are
predicted to further expand the capacity of the mRNA degradation code by
coupling it to dynamic events experienced by cells at specific
spatiotemporal coordinates within the developing embryo” (Alonso 2012).
As the preceding only begins to suggest, many wide-ranging factors bear on RNA degradation, and therefore on gene expression. Here we list some of them, but provide explanation only for those not described elsewhere in this document:
-
RNA decapping
“Eukaryotic mRNAs are post-transcriptionally modified, so that each
receives a 7-methylguanosine cap at its 5' end. The cap is joined to the
mRNA 5' end via a unique 5'-to-5' triphosphate linkage. The cap
distinguishes mRNAs from other RNA transcripts within the cell and
promotes splicing, export, translation and mRNA stability. Removal of the
cap is absolutely required before the mRNA can be digested by 5'-to-3'
exoribonucleases. Thus, decapping is a critical step in controlling mRNA
half-life and therefore gene expression”
(Coller 2016, doi:10.1038/nsmb.3315).
Decapping involves dynamic interactions among relevant factors.
“Decapping appears to involve a carefully orchestrated ‘dance’, in which
coactivators of decapping prime the [decapping] enzyme to bind to an
mRNA, close around the mRNA's 5' end and then create a composite active
site around the cap structure before cleavage. Why might such complexity
exist? Perhaps maintaining the decapping enzyme in a catalytically
inactive default state is critical to the cell — after all, so much is at
stake. The irreversible nature of the reaction might warrant a carefully
ordered set of steps to ensure that the timing is just right before the
mRNA is decapped and committed for destruction. Decapping is the ultimate
form of translational repression because it inhibits any further
translational initiation events and exposes the message to exonuclease
activity” (Coller 2016, doi:10.1038/nsmb.3315).
- RNA polyadenylation and deadenylation, and all the factors that bear on these processes. See “Alternative cleavage, polyadenylation, and deadenylation”.
- RNA polyuridylation See “RNA 3'-end oligouridylation” above.
-
Decay of mRNAs containing AU-rich elements (AREs)
“The canonical AREs generally have one or more copies of the AUUUA
pentamer that are usually embedded in a U-rich context. If they are
classified by sequence alone, canonical ARE-containing mRNAs constitute
up to 9% of cellular mRNAs. AREs have been grouped into three broad
categories based on the number and context of the AUUUA repeats”.
Decay begins with shortening of the polyadenylated tail of the
transcript, followed by degradation at both ends of the transcript by
exonucleases (Schoenberg and Maquat 2012).
- AREs are found in many types of transcript, including those that encode proto-oncogenes and those involved in the transition from cellular quiescence to proliferation. “It is not clear...why, under different conditions, some mRNAs with one or more AREs undergo accelerated decay and others do not” (Schoenberg and Maquat 2012).
- Proteins that bind to AREs “can be grouped by their stabilizing or destabilizing effects on the mRNA, by their type of RNA-binding motif and by the proteins that modify their action. ARE-BPs [ARE-binding proteins] can be regulated by kinases, phosphatases and, at least in one case, by an arginine methyltransferase. As a general rule [they] function as multimers and can be phosphorylated at the same site by different kinases or at different sites by different kinases” (Schoenberg and Maquat 2012).
- Nuclear receptors and mRNA decay. The glucocorticoid receptor, when bound by its ligand, not only indirectly affects ARE-mediated decay, but can itself directly bind to some mRNAs and activate their decay. “It remains to be determined how glucocorticoid receptor binding occurs and activates mRNA decay. Furthermore, it is not known whether ligand-dependent mRNA binding and destabilization is unique to the glucocorticoid receptor or whether it is also a property of other nuclear receptors” (Schoenberg and Maquat 2012).
- Decay of mRNAs containing GU-rich elements (GREs)
-
Nonsense-mediated mRNA decay (NMD)
“Nonsense-mediated mRNA decay (NMD) is arguably the best-studied
eukaryotic messenger RNA (mRNA) surveillance pathway, yet fundamental
questions concerning the molecular mechanism of target RNA selection
remain unsolved. Besides degrading defective mRNAs harboring premature
termination codons (PTCs), NMD also targets many mRNAs encoding
functional full-length proteins. Thus, NMD impacts on a cell’s
transcriptome and is implicated in a range of biological processes that
affect a broad spectrum of cellular homeostasis.”
“The current model posits that NMD is stimulated when the TC [termination codon] occurs in a microenvironment of the mRNP that is unfavorable for translation termination ... The majority of NMD-sensitive transcripts do not contain PTCs but are ordinary mRNAs coding for seemingly full-length functional proteins ... There is ample evidence that NMD can target both normal and erroneous transcripts. Among the NMD-inducing features, the presence of the 3′-most exon–exon junction >50 nt [nucleotides] downstream from the TC is the feature with the strongest predictive value for NMD susceptibility. PTCs resulting from mutations in the ORF [open reading frame] or from aberrant or alternative splicing, as well as genes with an intron in the 3′UTR mostly belong to this class of NMD targets. In addition, long 3′UTRs (>1000 nt in mammalian cells), the presence of actively translated short upstream ORFs (uORFs), or selenocysteine codons (UGA) in cells grown in the absence of selenium are also features that can — but not always do — trigger NMD. uORF translation often inhibits translation of the main ORF, either constitutively or in response to stress. Under such circumstances, ribosomes terminate at the TC of the uORF with usually several EJCs [exon junction complexes] remaining bound further downstream on the mRNA, which creates an NMD-promoting translation termination environment. Despite all these empirically determined features, it has so far remained impossible to computationally predict NMD targets with high confidence.” (Karousis and Mühlemann 2019, doi:10.1101/cshperspect.a032862)
“Nonsense-mediated mRNA decay (NMD), which is arguably the
best-characterized translation-dependent regulatory pathway in mammals,
selectively degrades mRNAs as a means of post-transcriptional gene
control. Control can be for the purpose of ensuring the quality of gene
expression. Alternatively, control can facilitate the adaptation of cells
to changes in their environment. The key to NMD, no matter what its
purpose, is the ATP-dependent RNA helicase upstream frameshift 1 (UPF1),
without which NMD fails to occur. However, UPF1 does much more than
regulate NMD. As examples, UPF1 is engaged in functionally diverse mRNA
decay pathways mediated by a variety of RNA-binding proteins that include
staufen, stem–loop-binding protein, glucocorticoid receptor, and regnase
1. Moreover, UPF1 promotes tudor-staphylococcal/micrococcal-like
nuclease-mediated microRNA decay. ... UPF1, as a protein polymath,
engenders cells with the ability to shape their transcriptome in response
to diverse biological and physiological needs”
(Kim and Maquat 2019, doi:10.1261/rna.070136.118).
- In mammals, nonsense-mediated mRNA decay is thought to apply only to newly synthesized transcripts during their pioneer round of translation — and particularly to transcripts containing premature stop codons, which would presumably lead to defective proteins if the transcripts were translated. (I believe a very recent study — spring or summer of 2013 — has shown that NMD can occur also in subsequent rounds of translation.)
- A substantial number of functional transcripts are downregulated by NMD, “suggesting that NMD functions in cellular processes in addition to quality control” (Schoenberg and Maquat 2012).
- “The recent proposal that NMD targets are the primary source of antigenic peptides for the major histocompatibility class I pathway, which presents endogenous cellular peptides to T cells, provides another emerging role for NMD in humans and, most likely, in all mammals” (Schoenberg and Maquat 2012).
- “The discovery that NMD is not only an RNA surveillance pathway but a regulator of normal gene expression has raised the possibility that NMD regulates normal biological processes, including development”. And, indeed, it appears that NMD “is critical for the differentiation of embryonic stem cells” — a role it plays by reducing mRNA levels for key pluripotency genes (Lou, Shum and Wilkinson 2015, doi:10.15252/embj.201591631).
- Interaction with alternative splicing: There is “a mechanism underlying the regulation of specific genes during development whereby a class of alternative exons that introduce a premature termination codon and activate nonsense-mediated mRNA decay are included [via alternative splicing] in adult tissues to suppress mRNA expression, but are skipped in embryonic tissues to activate mRNA expression” (Barash, Calarco, Gao et al. 2010).
- “Components of the nonsense-mediated mRNA decay (NMD) pathway have been implicated in regulating embryonic stem cell (ESC) differentiation, but the exact mechanism is unclear. Here we show that NMD controls expression levels of the translation initiation factor Eif4a2 and its premature termination codon-encoding isoform (Eif4a2PTC). NMD deficiency leads to translation of the truncated eIF4A2PTC protein. eIF4A2PTC elicits increased mTORC1 activity and translation rates and causes differentiation delays. This establishes a previously unknown feedback loop between NMD and translation initiation. Furthermore, our results show a clear hierarchy in the severity of target deregulation and differentiation phenotypes between NMD effector KOs (Smg5 KO > Smg6 KO > Smg7 KO), which highlights heterodimer-independent functions for SMG5 and SMG7. Together, our findings expose an intricate link between mRNA homeostasis and mTORC1 activity that must be maintained for normal dynamics of cell state transitions” (Huth, Santini, Galimberti et al. 2022, doi:10.1101/gad.347690.120).
-
Regulation of nonsense-mediated mRNA decay. An elaborate
array of proteins associated with an mRNA engage in an intricately
choreographed performance in order to perform the decay function.
Modifications of these proteins, and the signaling pathways that
produce these modifications or otherwise affect the proteins, play
regulatory roles that are too complex to detail here.
- One example of the larger picture: various stress conditions in a cell downregulate translation in general, and therefore also NMD. However (taking the example of stress due to hypoxia), the inhibition of NMD occurs only during the early stages of hypoxia, whereas normal translation is inhibited throughout the persistence of the condition. “This may be beneficial because several mRNAs that contribute to the cellular response to stress are NMD targets” — targets that, while toxic in unstressed cells and therefore repressed by NMD, are useful in stressed cells (Schoenberg and Maquat 2012).
- “Intriguingly, most mRNAs coding for NMD factors were among the NMD-sensitive transcripts” (Yepiskoposyan, Aeschimann, Nilsson et al. (2011). In other words, the mRNAs leading to the proteins carrying out nonsense-mediated mRNA decay are themselves subject to decay by these NMD proteins.
- “NMD can be regulated at the level of individual NMD factors or in feeback loops that coordinately control the abundance of one or more different NMD factors. ... Phosphorylation of NMD factors is an important mode of regulation of their activity and thus of NMD...The efficiency of NMD is also affected by microRNAs” (Schoenberg and Maquat 2012).
- NMD is also regulated based on tissue type and developmental stage.
- “Nonsense-mediated mRNA decay (NMD) is a surveillance pathway that recognizes and selectively degrades mRNAs carrying premature termination codons (PTCs). The level of sensitivity of a PTC-containing mRNA to NMD is multifactorial. We have previously shown that human β-globin mRNAs carrying PTCs in close proximity to the translation initiation AUG codon escape NMD. This was called the ‘AUG-proximity effect’. The present analysis of nonsense codons in the human α-globin mRNA illustrates that the determinants of the AUG-proximity effect are in fact quite complex, reflecting the ability of the ribosome to re-initiate translation 3' to the PTC and the specific sequence and secondary structure of the translated open reading frame. These data support a model in which the time taken to translate the short open reading frame, impacted by distance, sequence, and structure, not only modulates translation re-initiation, but also impacts on the exact boundary of AUG-proximity protection from NMD” (Pereira, Teixeira, Kong et al. 2015, doi:10.1093/nar/gkv588).
- No-go mRNA decay. Transcripts that stall on the ribosome during translation are degraded via endonucleolytic decay. Not much is known about the details.
- Non-stop mRNA decay. Transcripts lacking a stop codon are also degraded. Not much is known about the details.
- Promoter-mediated RNA degradation. The degradation of RNAs may depend in part on the interaction of proteins with the promoters of the genes from which the RNAs were transcribed or with other proteins bound to those promoters. One result of this interaction, for example, appears to be that a specific protein will accompany the RNA into the cytoplasm and play a role in the timing of the RNA’s degradation. (See the original research cited in Burgess 2012.)
- Alternatively spliced RNAs result (it’s not yet known how) in different lifespans for the RNA isoforms.
- Degradation by microRNAs and siRNAs. See “MicroRNA (miRNA) activity” and “Small interfering RNAs (siRNAs)”.
- Pseudogenes whose mRNA transcripts supply decoy targets for miRNAs that would otherwise target the corresponding normal mRNA. See “Pseudogenes”
- Antisense RNAs can bind to RNA transcripts in such a way as to either create a target site for an enzyme that will cleave (degrade) the transcript, or else block such a site, preventing cleavage.
-
Glucocorticoid receptor-mediated RNA decay
- “Glucocorticoid receptor (GR) has been shown recently to bind a subset of mRNAs and elicit rapid mRNA degradation ... Here, we demonstrate that GMD [glucocorticoid receptor-mediated mRNA decay] triggers rapid degradation of target mRNAs in a translation-independent and exon junction complex-independent manner, confirming that GMD is mechanistically distinct from nonsense-mediated mRNA decay (NMD). Efficient GMD requires PNRC2 (proline-rich nuclear receptor coregulatory protein 2) binding, helicase ability, and ATM-mediated phosphorylation of UPF1 (upstream frameshift 1). We also identify two GMD-specific factors: an RNA-binding protein, YBX1 (Y-box-binding protein 1), and an endoribonuclease, HRSP12 (heat-responsive protein 12). In particular, using HRSP12 variants, which are known to disrupt trimerization of HRSP12, we show that HRSP12 plays an essential role in the formation of a functionally active GMD complex. Moreover, we determine the hierarchical recruitment of GMD factors to target mRNAs. Finally, our genome-wide analysis shows that GMD targets a variety of transcripts, implicating roles in a wide range of cellular processes, including immune responses” (Park, Park, Yu et al. 2016, doi:10.1101/gad.286484.116).
-
Staufen1-mediated RNA decay
- The Staufen1 (Stau1) RNA-binding protein binds to certain sequences in the 3' untranslated regions of translationally active and folded messenger RNAs (mRNAs), leading to degradation of the mRNAs (Gong and Maquat 2011). However, Ricci, Kucukural, Cenik et al. (2014) found no evidence for mRNA degradation related to Stau1 binding in 3' UTRs.
- In a separate process, Staufen1 cooperates with cytoplasmic long, noncoding RNAs in degrading mRNAs. An Alu retrotransposon element in the cytoplasmic ncRNA imperfectly binds with an Alu element in the 3' untranslated region of the target mRNA. This combination in turn provides a binding site for the Staufen1 protein, again leading to degradation of the mRNA (Gong and Maquat 2011).
-
Intron retention. Certain (presynaptic) proteins are detectably
expressed in non-neuronal cells as well as neuronal ones. In the
former cells, a regulatory protein prevents the splicing out of a
3'-terminal intron, with the result that the incompletely spliced mRNA
is degraded in the nucleus (via a process involving the exosome
complex) and not exported into the cytoplasm. However, when expression
of the regulatory protein “decreases during neuronal differentiation,
the regulated introns are spliced out, thus allowing the accumulation
of translation-competent mRNAs in the cytoplasm”. In this way the
neuron-specific genes are expressed only in the appropriate context
(Yap, Lim, Khandelia et al. 2012).
“Recent landmark discoveries have underpinned the physiological
importance of intron retention across multiple domains of life and
revealed an unexpected breadth of functions in a large variety of
biological processes”
(Wong and Schmitz 2022, doi:10.1016/j.tig.2022.03.017).
- Factors supporting both decay and transcription. “Working in yeast, Haimovich et al. found that components of the cytoplasmic 5′ to 3′ decay pathway (collectively known as the 'decaysome') shuttle between the cytoplasm and nucleus. In the nucleus, they preferentially associate with chromatin near transcription start sites. The authors show that these factors stimulate transcription initiation and elongation and thus link transcription and decay” (Muers 2013). In other words, the same factors can support transcription and decay of mRNAs.
-
Codon Optimality
- Much of the foregoing relates to the decay of mRNAs considered “aberrant”. However: “We find here that codon usage within normal mRNAs also influences translating ribosomes and can have profound effects on mRNA stability”. “Genome-wide RNA decay analysis revealed that stable mRNAs are enriched in codons designated optimal, whereas unstable mRNAs contain predominately non-optimal codons. Substitution of optimal codons with synonymous, non-optimal codons results in dramatic mRNA destabilization” (Presnyak, Alhusaini, Chen et al. 2015, doi:10.1016/j.cell.2015.02.029).
- Consider two facts: (1) “We show that optimal codon content accounts for the similar stabilities observed in mRNAs encoding proteins with coordinated physiological function”; (2) “Recent studies reveal that tRNA concentrations within the cell are not static but are constantly undergoing change, sometimes dramatically. For instance ... tRNA concentrations vary widely between proliferating and differentiating cells”. Putting these two facts together: “Changes in cellular growth conditions and nutrient availability could significantly impact individual (or subsets of) charged tRNA levels. As a consequence of this reduction in supply, translational elongation rates of mRNAs enriched in the codons decoded by these tRNAs would be slowed and their levels decreased, due to enhanced turnover. In this way, codon optimality provides the cell not only with a general mechanism to hone mRNA levels but also with a mechanism to sense environmental conditions and rapidly tailor global patterns of gene expression”. “Based on our analysis, we would argue that significant alterations in tRNA concentrations could alter the mRNA expression profile within a cell by dynamically changing mRNA stability, even without any changes in transcription” (Presnyak, Alhusaini, Chen et al. 2015, doi:10.1016/j.cell.2015.02.029).
-
• “Synonymous codons are used non-randomly in the transcriptome
to shape multiple aspects of translation.
• “Optimal codons are associated with more efficient translation and correspond to cognate tRNA species that are more abundant and that are readily accommodated by the ribosome during translation.
• “The use of non-optimal codons can influence protein production by reducing ribosome translocation rates and causing ribosome collisions that can feed back to the translation initiation site.
• “Conserved, specific patterns of optimal and non-optimal codon use help to guide efficient co-translational folding and to minimize errors in translation.
• “Codon usage affects mRNA stability, and codon-influenced elongation stalling is sensed by the DEAD-box helicase Dhh1, which mediates codon-dependent variation in mRNA stability.
• “The interdependence between variable codon usage and the composition, charge status and post-transcriptional modifications of the tRNA pool enables global control of translation, which can be used to shape protein production to favour specific cellular programmes and to maintain homeostasis in conditions of stress or changes in nutritional status” (Hanson and Coller 2018, doi:10.1038/nrm.2017.91).
-
Co-translational mRNA decay.
- During translation, there are commonly multiple ribosomes along the length of an mRNA, with each ribosome producing a protein from the mRNA sequence. You can think of it as an mRNA passing, 5'-end first, through an array of ribosomes. The reference in the following quote to “the last translating ribosome” pertains to the last ribosome in the series: “It is generally assumed that mRNAs undergoing translation are protected from decay. Here, we show that mRNAs are, in fact, co-translationally degraded. This is a widespread and conserved process affecting most genes, where 5'–3' transcript degradation follows the last translating ribosome” (Pelechano, Wei and Steinmetz 2015, doi:10.1016/j.cell.2015.05.008).
-
Exosome
The exosome is a complex of proteins occurring in both the cell nucleus
and cytoplasm. It is “the most versatile RNA-degradation machine in
eukaryotes. The exosome has a central role in several aspects of RNA
biogenesis, including RNA maturation and surveillance. Moreover, it is
emerging as an important player in regulating the expression levels of
specific mRNAs in response to environmental cues and during cell
differentiation and development. Although the mechanisms by which RNA is
targeted to (or escapes from) the exosome are still not fully understood,
general principles have begun to emerge ... In addition, [there are]
previously unappreciated functions of the nuclear exosome, including in
transcription regulation and in the maintenance of genome stability”
(Kilchert, Wittmann and Vasiljeva 2016, doi:10.1038/nrm.2015.15).
- “The nuclear RNA exosome complex is involved in 3' processing of various stable RNA species and is crucial for RNA quality control in the nucleus. It also degrades many types of cryptic transcripts that are generated as a result of pervasive transcription and removes aberrant RNA molecules that failed to mature properly. Disruption of the RNA exosome or its cofactors is associated with human diseases” (Kilchert, Wittmann and Vasiljeva 2016, doi:10.1038/nrm.2015.15).
- “Targeting substrates to the exosome complex for degradation constitutes a two-step process. Exosome specificity factors recognize and bind to certain features on the target RNA and recruit activating complexes. Unwinding of the RNA substrate by helicases associated with the activating complexes facilitates RNA degradation by the exosome complex” (Kilchert, Wittmann and Vasiljeva 2016, doi:10.1038/nrm.2015.15).
- “Lack of proper mRNA processing that results in intron retention, transcription read-through or incorrect assembly of ribonucleoprotein particles (mRNPs) in the absence of packaging factors induces transcript degradation by the exosome complex” (Kilchert, Wittmann and Vasiljeva 2016, doi:10.1038/nrm.2015.15).
- “RNA surveillance by the exosome cooperates with RNA processing to regulate mRNA levels. Both the induction of non-productive RNA processing (such as premature transcription termination or cryptic splicing) or the suppression of proper mRNA processing (resulting in intron retention or read-through transcription) can be coupled with RNA decay by the exosome complex, thus reducing mRNA levels in response to external cues” (Kilchert, Wittmann and Vasiljeva 2016, doi:10.1038/nrm.2015.15).
- “Several novel functions that have been attributed to the exosome complex include the disassociation of stalled RNA polymerase II and resolving the formation of RNA–DNA hybrids, which are a source of genomic instability. These additional functions seem to be required to enable certain biological processes, such as the DNA damage response and antibody class switch recombination” (Kilchert, Wittmann and Vasiljeva 2016, doi:10.1038/nrm.2015.15).
- Supporting roles. Various enzymes and other molecules, each with its own regulatory context, play a role in the different RNA degradation processes. For example, there are at least two decapping enzymes in mammalian cells; they remove the “cap” from the 5' end of mRNA molecules, preparing the way for one sort of degradation. “mRNA decapping is a crucial step in the regulation of mRNA stability and gene expression” (Li, Song and Kiledjian 2011). We do not try to detail the many supporting molecules here. Nevertheless, these are not mere minor details; rather, they exemplify how processes of gene regulation permeate the entire organism, which is to say: how the organism employs its diverse powers in order to incorporate its DNA into the life of the whole.
In sum ... There are “complicated, multifactorial webs of regulatory events that coordinate the half-lives of cellular mRNAs, depending on the stage of organismal development, the type of tissue and the surrounding environmental conditions”. And a caveat: “Although mRNAs largely function to produce proteins, there is growing support for the idea that they can also serve as sinks for regulatory proteins and antisense ncRNAs such as miRNAs by functioning as ‘competing endogenous RNAs’ [see “Competing endogenous RNAs” above]. This indicates that the regulation of mRNA decay may cast a very broad net and affect as yet unappreciated cellular processes” (Schoenberg and Maquat 2012).
-
RNA decapping
“Translational control contributes immensely to the establishment of the
intricate complexity of genetic regulation that is necessary for the
development of multicellular organisms. It provides possibilities for
controlling the spatial deployment of a protein that cannot be achieved through
controlling transcription alone. Many translationally regulated mRNAs encode
proteins whose correct distributions are essential for developmental processes,
such as embryonic patterning, or for cellular processes, such as synaptic
transmission. Translational regulation of pre-existing mRNA can also provide
a highly dynamic temporal response, which is exemplified by the immediate
commencement after fertilization of global protein synthesis from maternally
expressed and silenced mRNAs in the embryos of many species. The central role
of post-transcriptional mechanisms of genetic regulation, including that of
translational control, in establishing the proteome and enabling cellular and
developmental processes is exemplified by the observation that only 40% of
variability of protein levels in mouse embryonic fibroblasts is attributable to
mRNA levels” (Kong and Lasko 2012).
“While most loss-of-function variants are rare, a subset have risen to high
frequency and occur in a homozygous state in healthy individuals. It is unknown
why these common variants are well tolerated, even though some affect essential
genes implicated in Mendelian disease ... Many common nonsense variants do not
ablate protein production from their host genes. We provide computational and
experimental evidence for diverse mechanisms of gene rescue, including
alternative splicing, stop codon readthrough, alternative translation
initiation, and C-terminal truncation. Our results suggest a molecular
explanation for the mild fitness costs of many common nonsense variants and
indicate that translational plasticity plays a prominent role in shaping human
genetic diversity” (Jagannathan and Bradley 2016, doi:10.1101/gr.205070.116 ).
“There is [now] a deeper appreciation that the [translational] mechanisms and
pathways of textbooks are only true in some circumstances, and that the
differing contexts of disease, development, and even subcellular location can
rewrite these mechanisms in interesting and surprising ways. For example, in
this issue Michal Minczuk and colleagues meticulously describe the many
distinctions between mitochondrial gene expression and nuclear gene expression,
including the unique structure of the mitochondrial ribosome and the mechanisms
that coordinate mitochondrial and cytosolic translation of the components of
the oxidative phosphorylation machinery. A provocative review by Christian
Spahn and colleagues discusses how viruses, and remarkably some eukaryotic
mRNAs, can use internal RNA structures (IRESs [internal ribosome entry sites])
to bypass traditional translation initiation at the 5'-end, first capturing and
then manipulating the eukaryotic translation machinery through non-canonical
interactions ... Moreover, there is increasing interest in how mRNA
modifications, structure, and binding proteins affect translation in a
spatiotemporal manner” (Neuman 2017, doi:10.1016/j.tibs.2017.06.003).
“Regulation of mRNA translation offers the opportunity to diversify the
expression and abundance of proteins made from individual gene products in
cells, tissues and organisms. Emerging evidence has highlighted variation in
the composition and activity of several large, highly conserved translation
complexes as a means to differentially control gene expression. Heterogeneity
and specialized functions of individual components of the ribosome and of the
translation initiation factor complexes eIF3 and eIF4F, which are required for
recruitment of the ribosome to the mRNA 5′ untranslated region, have been
identified. [There is] evidence for selective mRNA translation by components of
these macromolecular complexes as a means to dynamically control the
translation of the proteome in time and space”
(Genuth and Barna 2018, doi:10.1038/s41576-018-0008-z).
“The annotation of the mammalian protein-coding genome is incomplete. Arbitrary
size restriction of open reading frames (ORFs) and the absolute requirement for
a methionine codon as the sole initiator of translation have constrained the
identification of potentially important transcripts with non-canonical
protein-coding potential. Here, using unbiased transcriptomic approaches in
macrophages that respond to bacterial infection, we show that ribosomes
associate with a large number of RNAs that were previously annotated as
‘non-protein coding’. Although the idea that such non-canonical ORFs can encode
functional proteins is controversial, we identify a range of short and
non-ATG-initiated ORFs that can generate stable and spatially distinct
proteins. Notably, we show that the translation of a new ORF ‘hidden’ within
the long non-coding RNA Aw112010 is essential for the orchestration of
mucosal immunity during both bacterial infection and colitis. This work expands
our interpretation of the protein-coding genome and demonstrates that
proteinaceous products generated from non-canonical ORFs are crucial for the
immune response in vivo. We therefore propose that the misannotation of
non-canonical ORF-containing genes as non-coding RNAs may obscure the essential
role of a multitude of previously undiscovered protein-coding genes in immunity
and disease”
(Jackson, Kroehling, Khitun et al. 2018, doi:10.1038/s41586-018-0794-7).
“Adequate reprogramming of cellular metabolism in response to stresses or
suboptimal growth conditions involves a myriad of coordinated changes that
serve to promote cell survival. As protein synthesis is an energetically
expensive process, its regulation under stress is of critical importance.
Reprogramming of messenger RNA (mRNA) translation involves well‐understood
stress‐activated kinases that target components of translation initiation
machinery, resulting in the robust inhibition of general translation and
promotion of the translation of stress‐responsive proteins. Translational
arrest of mRNAs also results in the accumulation of transcripts in cytoplasmic
foci called stress granules. Recent studies focus on the key roles of transfer
RNA (tRNA) in stress‐induced translational reprogramming. These include
stress‐specific regulation of tRNA pools, codon‐biased translation influenced
by tRNA modifications, tRNA miscoding, and tRNA cleavage. In combination,
signal transduction pathways and tRNA metabolism changes regulate translation
during stress, resulting in adaptation and cell survival. This review examines
molecular mechanisms that regulate protein synthesis in response to stress”
(Advani and Ivanov 2019, doi:10.1002/bies.201900009).
“After stress, which can be caused by cell‐cycle synchronization, global
translation is repressed, while translation of some transcripts is selectively
maintained or induced. During the different cell‐cycle phases, translation of a
number of different transcripts is selectively up‐ or downregulated, but the
overall translation activity does not change significantly” (TOC blurb for
Anda and Grallert 2019, doi:10.1002/bies.201900022).
“During mRNA translation, the genetic information stored in mRNA is translated
into a protein sequence. It is imperative that the genetic information is
translated with high precision. Surprisingly, however, recent experimental
evidence has demonstrated that translation can be highly heterogeneous, even
among different mRNA molecules derived from a single gene in an individual
cell; multiple different polypeptides can be produced from a single mRNA
molecule and the rate of translation can vary in both space and time”
(Sonneveld, Verhagen and Tanenbaum 2020, doi:10.1016/j.tcb.2020.04.008).
“Stem cells are characterized by their ability to self-renew and differentiate
into many different cell types. Research has focused primarily on how these
processes are regulated at a transcriptional level. However, recent studies
have indicated that stem cell behaviour is strongly coupled to the regulation
of protein synthesis by the ribosome ... Stem cells are characterized by low
global translation rates despite high levels of ribosome biogenesis. The
maintenance of pluripotency, the commitment to a specific cell fate and the
switch to cell differentiation depend on the tight regulation of protein
synthesis and ribosome biogenesis. Translation regulatory mechanisms that
impact on stem cell function include mTOR signalling, ribosome levels, and mRNA
and tRNA features and amounts”
(Saba, Liakath-Ali, Green and Watt 2021, doi:10.1038/s41580-021-00386-2).
(In mice:)
“Retinal development is tightly regulated to ensure the generation of
appropriate cell types and the assembly of functional neuronal circuitry
... We discover thousands of genes that have dynamic changes at the
translational level and pervasive translational regulation in a developmental
stage-specific manner with specific biological functions. We further identify
genes whose translational efficiencies are frequently controlled by changing
usage in upstream open reading frame during retinal development. These genes
are enriched for biological functions highly important to neurons, such as
neuron projection organization and microtubule-based protein transport.
Surprisingly, we discover hundreds of previously uncharacterized micropeptides,
translated from putative long non-coding RNAs and circular RNAs. We validate
their protein products in vitro and in vivo and demonstrate their potentials in
regulating retinal development. Together, our study presents a rich and complex
landscape of translational regulation and provides novel insights into their
roles during retinogenesis”
(Chen, Chen, Li et al. 2021, doi:10.1093/nar/gkab749).
“The gene expression pathway from DNA sequence to functional protein is not as
straightforward as simple depictions of the central dogma might suggest. Each
step is highly regulated, with complex and only partially understood molecular
mechanisms at play. Translation is one step where the “one gene–one protein”
paradigm breaks down, as often a single mature eukaryotic mRNA leads to more
than one protein product. One way this occurs is through translation
reinitiation, in which a ribosome starts making protein from one initiation
site, translates until it terminates at a stop codon, but then escapes normal
recycling steps and subsequently reinitiates at a different downstream site.
This process is now recognized as both important and widespread, but we are
only beginning to understand the interplay of factors involved in termination,
recycling, and initiation that cause reinitiation events. There appear to be
several ways to subvert recycling to achieve productive reinitiation, different
types of stresses or signals that trigger this process, and the mechanism may
depend in part on where the event occurs in the body of an mRNA”
(Sherlock, Galvis, Vicens et al. 2023; doi:10.1261/rna.079375.122).
“Messenger RNA (mRNA) stability and translational efficiency are two crucial
aspects of the post-transcriptional process that profoundly impact protein
production in a cell. While it is widely known that ribosomes produce proteins,
studies during the past decade have surprisingly revealed that ribosomes also
control mRNA stability in a codon-dependent manner, a process referred to as
codon optimality. Therefore, codons, the three-nucleotide words read by the
ribosome, have a potent effect on mRNA stability and provide cis-regulatory
information that extends beyond the amino acids they encode. While the codon
optimality molecular mechanism is still unclear, the translation elongation
rate appears to trigger mRNA decay. Thus, transfer RNAs emerge as potential
master gene regulators affecting mRNA stability”
(Wu and Bazzini 2023; doi:10.1146/annurev-biochem-052621-091808).
“The application of ribosome profiling has revealed an unexpected abundance of
translation in addition to that responsible for the synthesis of previously
annotated protein-coding regions. Multiple short sequences have been found to
be translated within single RNA molecules, within both annotated protein-coding
and noncoding regions. The biological significance of this translation is a
matter of intensive investigation. However, current schematic or
annotation-based representations of mRNA translation generally do not account
for the apparent multitude of translated regions within the same molecules.
They also do not take into account the stochasticity of the process that allows
alternative translations of the same RNA molecules by different ribosomes.
There is a need for formal representations of mRNA complexity that would enable
the analysis of quantitative information on translation and more accurate
models for predicting the phenotypic effects of genetic variants affecting
translation. To address this, we developed a conceptually novel abstraction
that we term ribosome decision graphs (RDGs). RDGs represent translation as
multiple ribosome paths through untranslated and translated mRNA segments. We
termed the latter “translons.” Nondeterministic events, such as initiation,
reinitiation, selenocysteine insertion, or ribosomal frameshifting, are then
represented as branching points. This representation allows for an adequate
representation of eukaryotic translation complexity and focuses on locations
critical for translation regulation”
(Tierney, Świrski, Tjeldnes et al. 2024, doi:10.1101/gr.278810.123).
-
Translation initiation
This should be a major section, but was only lately added. Searching in
this document for "eIF" will take you to some scattered references to
(eukaryotic) translation initiation factors.
There is a considerable number of translation initiation factors, and, in
varying combinations, at least a dozen of them interact with each other,
with the ribosomal subunits, and with any given mRNA to prepare the way for
translation. Their regulatory potentials are vast, and are continually
being elucidated in the literature.
The general picture: “The major challenges in studying translation
initiation structurally are the number of steps and the size of the
complexes involved and the intricate choreography of conformational changes
required to scan for the proper start codon and act on it. Some initiation
factors are multisubunit complexes, such as the ∼750 kDa mammalian
initiation factor 3 (eIF3) composed of 13 proteins. Assembly of the
translation initiation complex on the small 40S ribosomal subunit occurs in
a stepwise fashion. The 40S⋅eIF3 particle bound with eukaryotic initiation
factors eIF1, eIF1A, and eIF5 recruits the methionine initiator tRNA
(Met-tRNAi) attached to GTP-bound eIF2. This 43S pre-initiation complex then
associates with an mRNA whose 7-methylguanosine 5' cap has been recognized
by the eIF4F multiprotein factor. The 43S particle scans the mRNA in the
5'-to-3' direction until it finds a start codon in an appropriate sequence
context. Met-tRNAi then base pairs with the AUG codon in the P site of the
small subunit, forming the 48S initiation complex. Joining with the 60S
ribosomal subunit completes the formation of the 80S ribosome that is ready
to proceed with protein synthesis” (Korostelev 2014,
doi:10.1016/j.cell.2014.10.005).
“The vast majority of eukaryotic messenger RNAs (mRNAs) initiate translation
through a canonical, cap-dependent mechanism requiring a free 5′ end and 5′
cap and several initiation factors to form a translationally active
ribosome. Stresses such as hypoxia, apoptosis, starvation, and viral
infection down-regulate cap-dependent translation during which alternative
mechanisms of translation initiation prevail to express proteins required to
cope with the stress, or to produce viral proteins. The diversity of
noncanonical initiation mechanisms encompasses a broad range of strategies
and cellular cofactors. Herein, we provide an overview and, whenever
possible, a mechanistic understanding of the various noncanonical mechanisms
of initiation used by cells and viruses” (Kwan and Thompson 2019,
doi:10.1101/cshperspect.a032672).
“Cells utilize transcriptional and posttranscriptional mechanisms to alter
gene expression in response to environmental cues. Gene-specific controls,
including changing the translation of specific messenger RNAs (mRNAs),
provide a rapid means to respond precisely to different conditions. Upstream
open reading frames (uORFs) are known to control the translation of mRNAs.
Recent studies in bacteria and eukaryotes have revealed the functions of
evolutionarily conserved uORF-encoded peptides. Some of these uORF-encoded
nascent peptides enable responses to specific metabolites to modulate the
translation of their mRNAs by stalling ribosomes and through ribosome
stalling may also modulate the level of their mRNAs. In this review, we
highlight several examples of conserved uORF nascent peptides that stall
ribosomes to regulate gene expression in response to specific metabolites in
bacteria, fungi, mammals, and plants”
(Dever, Ivanov and Sachs 2020, doi:10.1146/annurev-genet-112618-043822).
“Recent findings position the eukaryotic translation initiation factor eIF4E
as a novel modulator of mRNA splicing, a process that impacts the form and
function of resultant proteins. eIF4E physically interacts with the
spliceosome and with some intron-containing transcripts implying a direct
role in some splicing events. Moreover, eIF4E drives the production of key
components of the splicing machinery underpinning larger scale impacts on
splicing. These drive eIF4E-dependent reprogramming of the splicing
signature. This work completes a series of studies demonstrating eIF4E acts
in all the major mRNA maturation steps whereby eIF4E drives production of
the RNA processing machinery and escorts some transcripts through various
maturation steps. In this way, eIF4E couples the mRNA
processing-export-translation axis linking nuclear mRNA processing to
cytoplasmic translation. eIF4E elevation is linked to worse outcomes in
acute myeloid leukemia patients where these activities are dysregulated”
(Borden 2023, doi:10.1002/bies.202300145).
- “Mutation or inactivation of eIF3 [eukaryotic translation initiation factor 3] subunits results in developmental defects in Caenorhabditis elegans and zebrafish. Furthermore, analyses of human tumours reveal that overexpression of eIF3 is linked to diverse cancers, including breast, prostate and oesophageal malignancies. The integral role of eIF3 during cellular differentiation, growth and carcinogenesis suggests that eIF3 might drive specialized translation” (Lee, Kranzusch and Cate 2015, doi:10.1038/nature14267).
- “Recent studies have highlighted that certain factors possess roles outside of their general functions in translation. For example, the ribosome mediates translational specificity during development and viral infection through the requirement for distinct ribosomal proteins. During canonical translation, eIF3 acts as a protein scaffold for initiation complex assembly. Our results now reveal a new paradigm for translational control, in which, in addition to this general function, eIF3 can act as both an activator and repressor of cap-dependent transcript-specific translation through direct binding to defined RNA structural elements (Lee, Kranzusch and Cate 2015, doi:10.1038/nature14267).
- “[The RNA-binding protein] YTHDF1 recruits m6A-modified transcripts to facilitate translation initiation. The association of YTHDF1 with translation initiation machinery may be dependent on the loop structure mediated by eIF4G and the interaction of YTHDF1 with eIF3” (Wang, Zhao, Roundtree et al. 2015, doi:10.1016/j.cell.2015.05.014). Regarding the role of m6A-modified transcripts, see mRNA adenosine methylation above.
- “Our findings introduce the notion that cells harbor a distinct translation initiation pathway to respond to a variety of environmental conditions and cellular dysfunction. We showed that cells utilize a distinct, eIF2A-mediated initiation pathway, which includes uORF [upstream open reading frame] translation, to sustain expression of particular proteins [such as chaperone proteins] during the integrated stress response. [There are] thousands of predicted translation events in 5' UTRs [untranslated regions] and other noncoding RNAs ... Our observations underscore the importance of translation outside of annotated CDSs [protein coding sequences] and challenge the very definition of the U in 5′ UTR” (Starck, Tsai1, Chen et al. 2016, doi:10.1126/science.aad3867).
- “The eukaryotic translation initiation factor 4F (eIF4F) has become essentially synonymous with 5' cap-dependent mRNA translation. Recent studies demonstrate that cells assemble variants of eIF4F to produce adaptive, cap-dependent translatomes during physiological conditions that inhibit eIF4F ... So far, the evidence indicates that switching between eIF4Fs enables cells to reprogram their translational output (i.e., translatome), such that proteins that confer adaptive benefits are preferentially synthesized. Such translatome remodeling requires the interaction of eIF4Fs with RNA-binding proteins (RBPs), including RBM4 in the case of eIF4FH. These interactions represent a critical regulatory nexus that determines the translational priorities of mRNAs, especially given the multitude of RBPs and their complex relationships with other post-transcriptional regulators such as microRNAs. The identification of eIF4F variants that mediate the production of adaptive translatomes is in agreement with the recent appreciation that changes in translation efficiency, rather than mRNA concentration, is primarily responsible for stimulus-induced remodeling of the cellular proteome” (Ho and Lee 2016, 10.1016/j.tibs.2016.05.009).
-
The following from Hinnebusch 2017, doi:10.1016/j.tibs.2017.03.004:
“Initiation of translation on eukaryotic mRNAs generally follows the
scanning mechanism, wherein a preinitiation complex (PIC) assembled on
the small (40S) ribosomal subunit and containing initiator methionyl
tRNAi (Met-tRNAi) scans the mRNA leader for an AUG codon. In a current
model, the scanning PIC adopts an open conformation and rearranges to a
closed state, with fully accommodated Met-tRNAi, upon AUG recognition.
Evidence from recent high-resolution structures of PICs assembled with
different ligands supports this model and illuminates the molecular
functions of eukaryotic initiation factors eIF1, eIF1A, and eIF2 in
restricting to AUG codons the transition to the closed conformation. They
also reveal that the eIF3 complex interacts with multiple functional
sites in the PIC, rationalizing its participation in numerous steps of
initiation.
- “Recent high-resolution structures of PICs reveal distinct conformations of the 40S subunit, (initiator) tRNAi, and initiation factors indicative of different stages of the scanning mechanism for selecting AUG start codons.
- “An open PIC conformation features less tightly anchored mRNA and tRNAi, and unobstructed binding of the gatekeeper molecule eIF1 – all features compatible with scanning.
- “In the closed PIC conformation, both mRNA and tRNAi are locked into the decoding center, distorting eIF1 as a prelude to its release; and eIF1A stabilizes tRNAi binding – all compatible with AUG selection.
- “eIF2 subunits encase tRNAi within the TC; eIF2β helps to retain eIF1 in the open complex, and eIF2α interacts directly with ‘context’ mRNA nucleotides surrounding the AUG.
- “eIF3 effectively encircles the PIC and contacts various 40S functional sites, illuminating its multiple roles in stimulating PIC assembly, scanning, and AUG selection”.
- “A central mechanism regulating translation initiation in response to environmental stress involves phosphorylation of the α subunit of eukaryotic initiation factor 2 (eIF2α). Phosphorylation of eIF2α causes inhibition of global translation, which conserves energy and facilitates reprogramming of gene expression and signaling pathways that help to restore protein homeostasis. Coincident with repression of protein synthesis, many gene transcripts involved in the stress response are not affected or are even preferentially translated in response to increased eIF2α phosphorylation by mechanisms involving upstream open reading frames” (Wek 2018, doi:10.1101/cshperspect.a032870).
- “The conserved and essential DEAD-box RNA helicase Ded1p from yeast and its mammalian orthologue DDX3 are critical for the initiation of translation1 ... Here we show ... that the effects of Ded1p on the initiation of translation are connected to near-cognate initiation codons in 5′ untranslated regions. Ded1p associates with the translation pre-initiation complex at the mRNA entry channel and repressing the activity of Ded1p leads to the accumulation of RNA structure in 5′ untranslated regions, the initiation of translation from near-cognate start codons immediately upstream of these structures and decreased protein synthesis from the corresponding main open reading frames. The data reveal a program for the regulation of translation that links Ded1p, the activation of near-cognate start codons and mRNA structure. This program has a role in meiosis, in which a marked decrease in the levels of Ded1p is accompanied by the activation of the alternative translation initiation sites that are seen when the activity of Ded1p is repressed. Our observations indicate that Ded1p affects translation initiation by controlling the use of near-cognate initiation codons that are proximal to mRNA structure in 5′ untranslated regions.” (Guenther, Weinberg, Zubradt et al. 2018, doi:10.1038/s41586-018-0258-0)
- See also this item under “mRNA adenosine methylation”.
-
Translation speed and pausing
“Among the three phases of mRNA translation — initiation, elongation, and
termination — initiation has traditionally been considered to be rate
limiting and thus the focus of regulation. Emerging evidence, however,
demonstrates that control of ribosome translocation (polypeptide elongation)
can also be regulatory and indeed exerts a profound influence on
development, neurologic disease, and cell stress. The correspondence of mRNA
codon usage and the relative abundance of their cognate tRNAs is equally
important for mediating the rate of polypeptide elongation. [Recent
research shows] that ribosome pausing is a widely used mechanism for
controlling translation and, as a result, biological transitions in health
and disease” (Richter and Coller 2015, doi:10.1016/j.cell.2015.09.041).
The following abstract gives just a hint of the variety of factors involved
in the sort of pausing that yields productive elongation (I have yet to
extract all the relevant information from the paper):
“During protein synthesis, ribosomes encounter many roadblocks, the outcomes
of which are largely determined by substrate availability, amino acid
features and reaction kinetics. Prolonged ribosome stalling is likely to be
resolved by ribosome rescue or quality control pathways, whereas shorter
stalling is likely to be resolved by ongoing productive translation. How
ribosome function is affected by such hindrances can therefore have a
profound impact on the translational output (yield) of a particular mRNA. In
this Review, we focus on these roadblocks and the resumption of normal
translation elongation rather than on alternative fates wherein the stalled
ribosome triggers degradation of the mRNA and the incomplete protein
product. We discuss the fundamental stages of the translation process in
eukaryotes, from elongation through ribosome recycling, with particular
attention to recent discoveries of the complexity of the genetic code and
regulatory elements that control gene expression, including ribosome
stalling during elongation, the role of mRNA context in translation
termination and mechanisms of ribosome rescue that resemble recycling”
(Schuller and Green 2018, doi:10.1038/s41580-018-0011-4).
- “Numerous experiments have indicated that the speed and timings of translation may be critical to the formation of a protein’s native structure. For example...the removal of rare codons can reduce the specific activity of [the bacterial enzyme] chloramphenicol acetyltransferase...The rate of translation was recently demonstrated to affect the folding efficiency of Escherichia coli protein Suf1" (Saunders and Deane 2010).
- In mammalian cells, translational pausing was found to allow a nascent protein to “drag” the mRNA/ribosome/protein complex to the endoplasmic reticulum membrane, where the mRNA being translated undergoes efficient cytoplasmic splicing. Thus, both mRNA splicing and protein localization can be dependent upon translational pausing (Yanagitani, Kimata, Kadokura and Kohno 2011).
- Researchers found “universal patterns of conserved optimal and nonoptimal codons, often in clusters, which associate with the secondary structure of the translated polypeptides. ... These findings establish how mRNA sequences are generally under selection to optimze the cotranslational folding of corresponding polypeptides” (Pechmann and Frydman 2013). Optimal or nonoptimal codons consistently appear in “particular parts of the mRNA transcript, where they appear to strategically slow down or speed up translation. ‘What they are doing is setting a tune for protein folding’, said Frydman” (McClure 2013).
-
Role of the ribosome itself
“Like DNA, rRNA is extensively modified. Histones, once considered
as boring housekeeping proteins, are now clearly recognized as
active participants in chromatin remodelling and transcriptional
control through exquisite post-translational modifications
identified within histone tails. Likewise, the view of ribosomal
proteins as only carrying out rote-like functions is undergoing a
paradigm shift” (Xue and Barna 2012).
“Ribosomes are generally thought of as molecular machines with a
constitutive rather than regulatory role during protein synthesis. A study
by Slavov et al. now shows that ribosomes of distinct composition and
functionality exist within eukaryotic cells, giving credence to the concept
of ‘specialized’ ribosomes” (journal blurb for Preiss 2016,
doi:10.1016/j.tibs.2015.11.009).
“Ribosomes, the molecular machines behind translation, were once considered
to be an invariant driving force behind protein expression. However,
studies over the past decade paint a rather different picture; namely, that
ribosomes constitute an additional layer of regulatory control that might
define which subsets of mRNAs are translated, to what extent, and to what
purpose.”
“The work of Silver and colleagues (2007) was perhaps the first in-depth
demonstration that paralogous RPs [ribosomal proteins] can be functionally
distinct and exhibit specific effects on gene expression. While questions
abound regarding the mechanism, the works described here further implicate
RP specificity and, perhaps, RAPs [ribosome-associated proteins] in the
translational control of subsets of mRNAs in eukaryotes”
(Gerst 2018, doi://10.1016/j.tig.2018.08.004).
“The correct folding and processing of nascent polypeptides requires
ribosome-associated chaperones. One such chaperone, the ribosome-bound
nascent polypeptide–associated complex (NAC), cross-links to newly assembled
polypeptides. Gamerdinger et al. discovered that NAC is positioned above the
ribosomal exit site, from where it antagonizes incorrect endoplasmic
reticulum protein targeting. Remarkably, the extended N-terminal tail of the
β subunit inserts deeply inside the ribosomal tunnel to facilitate their
folding and sorting. As the peptide elongates, it displaces NAC from the
ribosomal tunnel. NAC then rearranges on the surface of the ribosome, ready
to coordinate further cotranslational activities”
(blurb in Science regarding this article:
Molecular Cell doi:10.1016/j.molcel.2019.06.030).
“Visualizing siRNA targeting of single mRNAs in living cells reveals that
passing ribosomes temporarily unfold the mRNA, exposing it to siRNA
recognition. This effect is due to the slow reorganization of many weak,
suboptimal interactions within the mRNA ... Variable and slow reorganization
of suboptimal RNA structures can explain why siRNA target sites remain open
for 30–60 seconds after a ribosome has left. The results of Ruijtenberg et
al. show that target site masking comes from multiple weak intramolecular
interactions, which occur over hundreds of base pairs ... Much remains to be
learnt about how to get the timing [of the unfolding of the mRNA] right.
(Małecka and Woodson 2020, doi:10.1038/s41594-020-0495-4).
“A universal property of all rRNAs explored to date is the prevalence of
post-transcriptional (“epitranscriptional”) modifications, which expand the
chemical and topological properties of the four standard nucleosides. Are
these modifications an inert, constitutive part of the ribosome? Or could
they, in part, also regulate the structure or function of the ribosome? In
this review, we summarize emerging evidence that rRNA modifications are more
heterogeneous than previously thought, and that they can also vary from one
condition to another, such as in the context of a cellular response or a
developmental trajectory”
(Georgeson and Schwartz 2021, doi:10.1261/rna.078859.121).
“We used RiboMeth-seq to demonstrate that differential 2′-O-methylation of
ribosomal RNA (rRNA) represents a considerable source of ribosome
heterogeneity in human cells, and that modification levels at distinct sites
can change dynamically in response to upstream signaling pathways, such as
MYC oncogene expression. Ablation of one prominent methylation resulted in
altered translation of select mRNAs and corresponding changes in cellular
phenotypes. Thus, differential rRNA 2′-O-methylation can give rise to
ribosomes with specialized function. This suggests a broader mechanism where
the specific regulation of rRNA modification patterns fine tunes
translation”
(Jansson, Häfner, Altinel et al. 2021, doi:10.1038/s41594-021-00669-4).
- “Emerging studies reveal that ribosome activity may be highly regulated. Heterogeneity in ribosome composition resulting from differential expression and post-translational modifications of ribosomal proteins, ribosomal RNA (rRNA) diversity and the activity of ribosome-associated factors may generate ‘specialized ribosomes’ that have a substantial impact on how the genomic template is translated into functional proteins. Moreover, constitutive components of the ribosome may also exert more specialized activities by virtue of their interactions with specific mRNA regulatory elements such as internal ribosome entry sites (IRESs) or upstream open reading frames (uORFs). Here we discuss the hypothesis that intrinsic regulation by the ribosome acts to selectively translate subsets of mRNAs harbouring unique cis-regulatory elements, thereby introducing an additional level of regulation in gene expression and the life of an organism” (Xue and Barna 2012).
- One particular ribosomal protein (RPL38) has recently been shown to help determine which mRNAs are preferentially translated (in a tissue-specific manner) by the ribosome it is associated with. RPL38 seems to be particularly connected to gene expression during embryonic development (Kondrashov, Pusic, Stumpf et al. 2011; Topisirovic and Sonenberg 2011).
-
Translational recoding
“It is generally assumed that (1) all codons encode identical information
in all organisms (with few exceptions), and (2) the reading frame is
invariant. Beginning in the mid-1970s, mRNA elements were discovered that
direct ribosomes to reassign the meanings of codons, induce ribosomes to
slip into alternative reading frames ( programmed ribosomal frameshifting
[PRF]), and even bypass long stretches of mRNA sequence (ribosome shunting).
All of these were eventually subsumed under the general heading of
‘translational recoding,’ defined as instances in which ‘...the rules for
decoding are temporarily altered through the action of specific signals
built into the mRNA sequences’”
(Dever, Dinman and Green 2018, doi:10.1101/cshperspect.a032649).
Dever, Dinman and Green (preceding item) cite the following general classes
of event: (1) “recoding directed by ‘flat’ cis-acting sequence elements”;
(2) “recoding directed by cis-acting topological features” such as mRNA stem
loops and pseudo-knots; (3) “recoding directed by trans-acting factors” such
as small molecules, trans-acting proteins, and trans-acting nucleic acids.
-
Mitochondrial ribosomal protein binding to cytoplasmic ribosomes
“Mammalian cells have both cytoplasmic and mitochondrial ribosomes, which
have long been considered to operate completely independently. However, a
new report shows that after heat shock, MRPL18, a human mitochondrial
ribosomal protein, binds to cytoplasmic ribosomes to influence translation
of heat-shock mRNAs” (Warner 2015, doi:10.1038/nsmb.3023, reporting on work
by Zhang, Gao, Coots et al., doi:10.1038/nsmb.3000).
- In a human cell line: “in response to heat shock, the phosphorylation of translational initiation factor eIF2α causes cytoplasmic ribosomes ... to initiate translation of the mRNA encoding the mitochondrial ribosomal protein MRPL18 at an unusual CUG codon to generate the protein MRPL18(cyto), which lacks the mitochondrial signal sequence and remains in the cytoplasm. This truncated MRPL18(cyto) is itself phosphorylated by the heat shock–activated Lyn kinase, thus leading to its association with cytoplasmic ribosomes. These hybrid ribosomes are activated for cap-independent translation of mRNAs encoding heat-shock proteins such as HSP70 and HSP40. The physiological importance of this phenomenon is established by the observation that lack of MRPL18(cyto) prevents the thermotolerance that cells develop when initially exposed to a mild heat shock” (Warner 2015, doi:10.1038/nsmb.3023, reporting on work by Zhang, Gao, Coots et al., doi:10.1038/nsmb.3000).
- The foregoing discovery was rather dramatic and unexpected. Further, “in order to stimulate the translation of HSP70-encoding mRNAs under conditions in which translation of most mRNAs is reduced, the cell integrates, in a quite novel way, at least three types of translational controls that have been described in the past few decades”: (1) the presence of an upstream ORF [open reading frame] in combination with phosphorylation of a translation initiation factor leads to a slowing of translation initiation and the skipping of the “normal” start codon. The result is a protein shortened at the upstream end. Importantly, phosphorylation of the translation initiation factor can be performed in response to various kinds of stress in addition to heat shock, so that the kind of process recorded here presumably bears on multiple cellular conditions. (2) The shortened mitochondrial ribosomal protein, now bound to a cytoplasmic ribosome, yields a “hybrid” ribosome “essential for translation of the HSP70-encoding mRNA. The relatively new concept of 'specialized' ribosomes, tailored for the translation of specific mRNAs, has now been demonstrated in several instances and may prove to be an important element in the overall regulation of translation”. (3) The altered mitochondrial ribosomal protein, when bound to a cytoplasmic ribosome, “permits the hybrid ribosome to bypass normal cap-dependent initiation [thereby enabling translation of the stress-related HSP70-encoding mRNA], presumably by interacting with some structure in the 5' UTR to effect translation” (Warner 2015, doi:10.1038/nsmb.3023, reporting on work by Zhang, Gao, Coots et al., doi:10.1038/nsmb.3000).
-
RNA sequence
mRNA sequence, of course, has a bearing on many other topics in this
section. For example, RNA structure (see
RNA structure below) is intimately related
to sequence. Here we look at sequence issues not discussed elsewhere.
Recent work by Kwon et al. using reporter RNAs has “led to the discovery of
a GGC motif [in RNAs] that inhibits translation and an A-rich element that
promotes cap-independent translation. In both cases, mRNA stability is
altered as a consequence of altered translation. Examining the GGC element
in more detail, the authors concluded that it inhibits translation via
G-quadruplex formation, which is usually minimized in cells by the activity
of the helicase DHX36 ...
“The A-rich element that Kwon et al. discovered also highlights some emerging concepts. Firstly, although this element stabilizes mRNAs by stimulating translation, this process is relatively inefficient, and many such mRNA molecules do not engage ribosomes. For these non-translating mRNAs, the A-rich element actually leads to destabilization, which is in agreement with a previous study. Thus, the same element can both stabilize and destabilize an mRNA via mechanisms that are translation-dependent and translation-independent, respectively. Furthermore, stabilizing and destabilizing effects both depend on the poly(A)-binding protein PAPB1, demonstrating that readers of RNA regulatory elements also play opposing roles. Dualities such as this are a recurrent theme in post-transcriptional regulation and highlight how RNA fate decisions involve the integration of many signals and kinetic competition between translation and decay ...
“These results portray post-transcriptional regulation as relatively chaotic, with many sequence elements and readers, each performing diverse roles. However, cells do manage to impart some order ...
“The work of Kwon et al. and others also demonstrates that regulatory interactions occur along the entire length of an mRNA transcript, rather than being concentrated in a few regions. This raises questions of whether and how signals are interpreted differently when located in distinct regions of an mRNA or, indeed, within different mRNAs altogether” (Bühler and Tuck 2020, doi:10.1038/s41594-020-0482-9).
-
Small open reading frames and upstream open reading frames (uORF)
Thousands of small open reading frames have been discovered outside
annotated coding regions in recent years. “New studies over the past
decade have revealed the existence of multiple translated ORFs hidden
within the transcriptome, revealing an additional level of complexity in
the translatome that was previously unappreciated. Translated ORFs can
correspond to functional proteins, but also be of a regulatory nature ...
regulatory ORFs are usually small and located in regions generally
believed to be noncoding, such as 5' UTRs. They can modulate expression
of other proteins or RNAs in trans, by the action of the encoded
peptide, or in cis, by directly interfering with the expression of
a downstream ORF. The number of transcripts with potentially translated
uORFs is large, and future studies will be required to identify those
with the highest impact in translational regulation”
(Ruiz-Orera and Albà 2019, doi:10.1016/j.tig.2018.12.003).
The functions of micropeptides “are very heterogeneous, although several
of them have been shown to play roles in muscle function ... An
interesting case is the developmental gene Mlpt/Tal/Pri found in
different insect species. The gene was initially believed to be noncoding
but it was later discovered that it encoded several functional peptides.
These peptides, produced from a polycistronic transcript, direct the
proteolytic cleavage of the transcription factor Shavenbaby (Svb),
converting it from a repressor to an activator. Another example is the
gene MIEF1, which translates a protein involved in the regulation of
mitochondrial fission but also contains another ORF translating a
70-amino-acid micropeptide. The latter peptide is conserved across
vertebrates (MIEF1-MP) and regulates mitochondrial translation by binding
to the mitoribosome”
(Ruiz-Orera and Albà 2019, doi:10.1016/j.tig.2018.12.003).
Between a third and a half of human genes have one or more upstream open
reading frames — protein-coding sequences in the 5' untranslated region
(5' UTR), upstream from the main ORF. These can be extremely short
sequences, or more substantial, and their translation can repress the
translation of the main ORF. But many regulatory possibilities exist:
“There are numerous alternative mechanisms that control the synthesis of
a protein whose mRNA contains uORFs”. These include: length, secondary
structure, and GC content of the 5' UTR; where the uORF is located, its
distance from the mRNA cap, and the distance between the uORF termination
and the main ORF; presence of an AUG or non-AUG start codon; the strength
of the consensus initiation sequence of the uORF (“Kozak sequence”); the
length of the uORF; and the presence of an overlap between the uORF and
the main ORF (Summers, Pöyry and Willis 2013,
doi:10.1016/j.biocel.2013.04.020). Obviously, then, this section could
be greatly filled out.
-
An example: the ATF4 mRNA, which encodes a transcription factor, has
two uORFs in its 5' UTR. uORF1 is 3 amino acids long and uORF2, which
is 59 amino acids long, overlaps with the main ATF4 ORF. Under most
conditions uORF1 is efficiently translated, after which the
translating ribosome has time to reacquire a necessary translation
initiation factor before reaching the start codon of uORF2. Because
the longer uORF2 overlaps the main ORF, ribosomes commonly do not
translate the latter, so that production of ATF4 is effectively
repressed by the uORFs. However, during certain stress conditions, a
subunit of a translation initiation factor (eIF2) undergoes increased
phosphorylation, which reduces the concentration of a crucial
initiation complex. In this case the ribosome, probably not having
acquired the necessary initiation factors in time, will bypass the
uORF2 start codon, and therefore will be able to re-initiate
translation later, and further downstream, at the ATF4 ORF (Summers,
Pöyry and Willis 2013, doi:10.1016/j.biocel.2013.04.020).
And so increased phosphorylation of a translation initiation factor, which tends to reduce the concentration of that factor and thus repress the translation of many genes, has the opposite effect on genes such as ATF4.
- It’s been found that several codons other than the standard AUG start codon are used by the ribosome for beginning translation of uORFs, albeit at lower rates of translation. Some of these alternative start codons lead to production of peptides (short proteins). Translation from the weaker start codons is assumed to have correspondingly weaker regulatory functions than translation from AUG codons. A translation initiation factor (eIF1) plays a role in determining whether or not alternative start codons are employed in translation, with a high concentration of eIF1 abolishing initiation from non-AUG codons. eIF1 itself is presumably involved in a feedback loop affecting its own translation (Summers, Pöyry and Willis 2013, doi:10.1016/j.biocel.2013.04.020).
- “Translated uORFs correlate with repression of the downstream CDS [coding DNA sequence] translation. Moreover, overlapping open reading frames (oORFs) act as stronger repressors of CDS translation”. “Dynamic regulation of specific transcripts can result from the interaction between repressive uORFs and sequence‐specific RNA‐binding proteins” (McGeachy and Ingolia 2016, doi:10.15252/embj.201693946, reporting on work by Johnstone, Bazzini and Giraldez 2016, doi:10.15252/embj.201592759).
-
An example: the ATF4 mRNA, which encodes a transcription factor, has
two uORFs in its 5' UTR. uORF1 is 3 amino acids long and uORF2, which
is 59 amino acids long, overlaps with the main ATF4 ORF. Under most
conditions uORF1 is efficiently translated, after which the
translating ribosome has time to reacquire a necessary translation
initiation factor before reaching the start codon of uORF2. Because
the longer uORF2 overlaps the main ORF, ribosomes commonly do not
translate the latter, so that production of ATF4 is effectively
repressed by the uORFs. However, during certain stress conditions, a
subunit of a translation initiation factor (eIF2) undergoes increased
phosphorylation, which reduces the concentration of a crucial
initiation complex. In this case the ribosome, probably not having
acquired the necessary initiation factors in time, will bypass the
uORF2 start codon, and therefore will be able to re-initiate
translation later, and further downstream, at the ATF4 ORF (Summers,
Pöyry and Willis 2013, doi:10.1016/j.biocel.2013.04.020).
-
Internal ribosome entry sites (IRESs)
- “Emerging evidence suggests that the ribosome has a regulatory function in directing how the genome is translated in time and space. However, how this regulation is encoded in the messenger RNA sequence remains largely unknown. Here we uncover unique RNA regulons embedded in homeobox (Hox) 5' untranslated regions (UTRs) that confer ribosome-mediated control of gene expression. These structured RNA elements, resembling viral internal ribosome entry sites (IRESs), are found in subsets of Hox mRNAs. They facilitate ribosome recruitment and require the ribosomal protein RPL38 for their activity. Despite numerous layers of Hox gene regulation, these IRES elements are essential for converting Hox transcripts into proteins to pattern the mammalian body plan. This specialized mode of IRES-dependent translation is enabled by an additional regulatory element that we term the translation inhibitory element (TIE), which blocks cap-dependent translation of transcripts. Together, these data uncover a new paradigm for ribosome-mediated control of gene expression and organismal development” (Xue, Tian, Fujii et al. 2015, doi:10.1038/nature14010).
- “It will be interesting to determine if additional ribosomal proteins may promote specialized translation through control of unique subsets of IRES-containing mRNAs, either directly or through RNA-binding proteins. For example, RPS25 is required for IRES-dependent translation of certain viral IRES mRNAs. Moreover, rRNA modifications both at the level of pseudouridylation and RPL13a-dependent methylation also appear to regulate the translation of certain cellular IRES-containing mRNAs. We therefore speculate that similar to the complex and highly regulated system of transcriptional control, in which specific DNA sequences and histone marks regulate gene expression, cis-acting RNA regulons, in conjunction with more specialized ribosome activity, provide newfound regulatory control to gene expression critical for mammalian development” (Xue, Tian, Fujii et al. 2015, doi:10.1038/nature14010).
- “This study found that Lys109, Lys121 and Lys122 represent critical ubiquitination sites for far upstream element-binding protein 2 (KHSRP), a negative ITAF [IRES trans-acting factor]. Mutations at these sites subsequently reduced KHSRP ubiquitination and abolished its inhibitory effect on IRES-driven translation ... these results show that ubiquitination can exert control over IRES-driven translation via modification of ITAFs, and to the best of our knowledge, this is the first description of such a regulatory mechanism for IRES-dependent translation” (Kung, Hung, Chien and Shih 2017, doi:10.1093/nar/gkw1042).
-
Codon usage
- A team of researchers looking at the relation between mRNA degradation and the usage of optimal or non-optimal codons (see Codon optimality) cites the crucial role of the ribosome in “sensing” the character of the mRNA it is translating. Codon usage affects translocation of the mRNA through the ribosome, with profound consequences for mRNA stability. “The ribosome acts as the master sensor, helping to determine the fate of all mRNAs, both normal and aberrant, through modulation of its elongation and/or termination processes ... We suggest that a component of mRNA stability is built into all mRNAs as a function of codon composition. The elongation rate of translating ribosomes is communicated to the general decay machinery, which affects the rate of deadenylation and decapping” (Presnyak, Alhusaini, Chen et al. 2015, doi:10.1016/j.cell.2015.02.029).
- “The genetic code determines how amino acids are encoded within mRNA. It is universal among the vast majority of organisms, although several exceptions are known. Variant genetic codes are found in ciliates, mitochondria, and numerous other organisms. All revealed genetic codes (standard and variant) have at least one codon encoding a translation stop signal. However, recently two new genetic codes with a reassignment of all three stop codons were revealed in studies examining the protozoa transcriptomes. Here, we discuss this finding and the recent studies of variant genetic codes in eukaryotes. We consider the possible molecular mechanisms allowing the use of certain codons as sense and stop signals simultaneously. The results obtained by studying these amazing organisms represent a new and exciting insight into the mechanism of stop codon decoding in eukaryotes” (Alkalaeva and Mikhaileva 2017, doi:10.1002/bies.201600213).
- ““The genetic code, which defines the amino acid sequence of a protein, also contains information that influences the rate and efficiency of translation. Neither the mechanisms nor functions of codon-mediated regulation were well understood. The prevailing model was that the slow translation of codons decoded by rare tRNAs reduces efficiency. Recent genome-wide analyses have clarified several issues. Specific codons and codon combinations modulate ribosome speed and facilitate protein folding. However, tRNA availability is not the sole determinant of rate; rather, interactions between adjacent codons and wobble base pairing are key. One mechanism linking translation efficiency and codon use is that slower decoding is coupled to reduced mRNA stability. Changes in tRNA supply mediate biological regulation — for instance, changes in tRNA amounts facilitate cancer metastasis” (Brule and Grayhack 2017, doi:10.1016/j.tig.2017.02.001).
-
Small open reading frames and upstream open reading frames (uORF)
-
RNA structure
- Work on a bacterial gene has shown that the first 20 or so codons at the beginning (5’ end) of its mRNA must be maintained in a flexible (unstructured) manner in order to allow ribosome binding and translation initiation (Loh, Memarpour, Vaitkevicius et al. 2012).
- “Programmed −1 ribosomal frameshift (−1 PRF) signals redirect translating ribosomes to slip back one base on messenger RNAs. ... Here we describe a −1 PRF signal in the human mRNA encoding CCR5, the HIV-1 co-receptor. CCR5 mRNA-mediated −1 PRF is directed by an mRNA pseudoknot, and is stimulated by at least two microRNAs. Mapping the mRNA–miRNA interaction suggests that formation of a triplex RNA structure stimulates −1 PRF. A −1 PRF event on the CCR5 mRNA directs translating ribosomes to a premature termination codon, destabilizing it through the nonsense-mediated mRNA decay pathway. At least one additional mRNA decay pathway is also involved. Functional −1 PRF signals that seem to be regulated by miRNAs are also demonstrated in mRNAs encoding six other cytokine receptors, suggesting a novel mode through which immune responses may be fine-tuned in mammalian cells” (Belew, Meskauskas, Musalgaonkar et al. 2014).
- “Here we show that G-quadruplex RNA when introduced within coding regions are capable of stimulating –1 ribosomal frameshifting in vitro and in cultured [mammalian] cells. Systematic manipulation of the loop length between each G-tract revealed that the –1 frameshifting positively correlates with G-quadruplex stability. ... Further, we demonstrated that the G-quadruplexes can stimulate +1 frameshifting and stop codon readthrough as well. These results suggest a potentially novel translational gene regulation mechanism mediated by G4 RNA” (Yu, Teulade-Fichou and Olsthoorn 2014).
- “Programmed −1 ribosomal frameshift (−1 PRF) signals redirect translating ribosomes to slip back one base on messenger RNAs. ... Here we describe a −1 PRF signal in the human mRNA encoding CCR5, the HIV-1 co-receptor. CCR5 mRNA-mediated −1 PRF is directed by an mRNA pseudoknot, and is stimulated by at least two microRNAs. Mapping the mRNA–miRNA interaction suggests that formation of a triplex RNA structure stimulates −1 PRF. A −1 PRF event on the CCR5 mRNA directs translating ribosomes to a premature termination codon, destabilizing it through the nonsense-mediated mRNA decay pathway. At least one additional mRNA decay pathway is also involved. Functional −1 PRF signals that seem to be regulated by miRNAs are also demonstrated in mRNAs encoding six other cytokine receptors, suggesting a novel mode through which immune responses may be fine-tuned in mammalian cells” (Belew, Meskauskas, Musalgaonkar et al. 2014).
- See also RNA structure and dynamics under THREE-DIMENSIONAL ORGANIZATION OF CHROMOSOMES, NUCLEUS, AND CELL
-
Temperature-controlled translation
- Regarding the prfA gene in bacteria: “An RNA thermosensor located within the [prfA transcript’s 5’-untranslated region] obstructs binding of the ribosome at low temperatures. Second, a trans-acting riboswitch has been shown to down-regulate PrfA translation by binding to the thermosensor at higher temperatures (Loh, Memarpour, Vaitkevicius et al. 2012).
-
Transfer RNA (tRNA)
“Alterations in transcript‐specific translation are emerging as a driver of
cellular transformation and cancer etiology. A new study provides evidence
for enhanced codon‐dependent translation of hypoxia‐inducible factor 1α in
promoting glycolytic metabolism and drug resistance in melanoma cells. This
specialized translation reprogramming relies, in part, on mTORC2‐mediated
phosphorylation of enzymes modifying the wobble position of the transfer RNA
anticodon”. “These exciting findings ... open a new portal into
investigating the role of tRNA modifications and codon-mediated translation
regulation in cancer pathogenesis”.
(McMahon and Ruggero 2018, doi:10.15252/embj.201899978).
-
RNA-binding proteins
“A constellation of RNA-binding proteins exerts post-transcriptional
control over the fate of mRNA expression. The stability and capacity
of an mRNA to be translated are highly regulated through sequence
elements in the mRNA and the proteins that recognize them. Some
proteins...directly bind specific sequences, whereas others, such as
Argonaute (Ago) proteins, require small RNA guides for recruitment to
target sites. Individually, [such] proteins have been shown to
regulate the stability or translational efficiency of [ribosome-]bound
mRNAs (Pasquinelli 2012).
Mammalian ribosomes “associate with various accessory proteins, forming the
ribosome–protein interactome ... [Researchers] identified ~430
RAPs [ribosome-associated proteins] which fall into three categories:
proteins that are involved in mRNA modifications; enzymes that mediate
post-translational modifications (PTMs); and proteins that are implicated in
basic cellular functions, including the cell cycle, reduction-oxidation
(redox) homeostasis, and metabolism. ¶ One of the identified RAPs was UFL1,
which is an enzyme that mediates protein ufmylation — a metazoan-specific
post-translational modification that resembles ubiquitylation. Three
ribosomal proteins and one translation initiation factor were found to be
ufmylated, indicating that the association of UFL1 with ribosomes has
functional importance ... This study suggests that specific RAPs can bind to
a subset of ribosomes and regulate translation at defined subcellular
localizations. It thus reveals that the mammalian ribo-interactome is highly
complex and that RAPs may provide additional levels of translation
regulation (Strzyz 2017, doi:10.1038/nrm.2017.62).
Of the numerous RNA-binding proteins affecting translation directly or
indirectly, some are mentioned under other headings in this document.
-
“Meiotic progression is controlled by cytoplasmic polyadenylation and
translational activation of masked, maternal mRNAs. RNA-binding-protein
interactions with adjacent cis elements cause local conformational
changes to the mRNAs that determine the extent and timing of their
activation”. “Masked mRNAs have short poly(A) tails, whose extension can
be promoted by sequence-specific interactions between CPEB and the
cytoplasmic polyadenylation element (CPE), an RNA motif in the 3'
untranslated region (3' UTR). There are four CPEB proteins in
vertebrates. Phosphorylation of CPE-bound CPEB permits remodeling of the
RNP complex and promotes recruitment of a poly(A) polymerase that
elongates the tail of the mRNA. This in turn promotes recruitment of
translation initiation factors, thus activating translation”.
“CPE–CPEB mediated translational regulation is widespread. Computational analysis with some empirical validation indicates that as many as 20–40% of mRNAs in Xenopus and mammals, including humans, may be subject to such regulation. However, not all CPE-containing mRNAs are regulated in the same way. Some ’early’ mRNAs are polyadenylated and translationally activated at meiotic prophase I, whereas ‘late’ mRNAs are activated at metaphase I. Still, other CPE-containing mRNAs do not undergo translational regulation. Whether and how a particular CPE-containing mRNA is regulated depends not only on the CPE and its distance from the polyadenylation signal (PAS) but also on other cis-acting elements in its 3' UTR, leading to the concept of a ‘combinatorial code’ of regulatory motifs. Other relevant cis-acting elements include additional CPEs, AU-rich elements (AREs) and elements that bind the RNA-binding proteins Pumilio and Msi (PBEs and MBEs, respectively)”.
Among those other elements in Drosophila is Msi, containing (like CPEB) two RNA recognition motifs. Depending on context, Msi can act as a translation activator or repressor. It turns out that the nature of the interaction between CPEB and Msi affect whether an mRNA is translated. Most studies on translational regulation to date have focused on single molecules. But the suggestion now is that “cooperative interactions between CPEB1 and Msi1 are widespread”, and that interactions of this sort could come to bear on a substantial percentage of mRNAs (Lasko 2017, doi:10.1038/nsmb.3445).
-
Staufen1 (Stau1) protein
“Like many RNA-binding factors, the Drosophila and mammalian
Staufen proteins have been implicated in multiple
post-transcriptional processes including alternative splicing, RNA
localization, translational activation and translation-dependent
mRNA decay. Which activity is observed depends on the cellular
context, the identity of the bound RNA and the location of the
binding site on the target RNA” (Ricci, Kucukural, Cenik et al.
2014).
- The Staufen1 (Stau1) protein “interacts with actively translating ribosomes and with mRNA coding sequences (CDSs) and 3' UTRs in proportion to their GC content and propensity to form internal secondary structure. On mRNAs with high CDS GC content, higher Stau1 levels lead to greater ribosome densities, thus suggesting a general role for Stau1 in modulating translation elongation through structured CDS regions. Our results also indicate that Stau1 regulates translation of transcription-regulatory proteins” (Ricci, Kucukural, Cenik et al. 2014).
- In Drosophila, mRNAs encoding transcription-regulatory proteins were found to be enriched in Staufen contacts. In a study of human cells, a number of mRNAs encoding transcription factors were “highly enriched” for occupancy by Stau1 in both the 3' UTR and coding regions. “Transcription factors and zinc-binding proteins were also highly enriched among the mRNAs whose ribosome density was most positively affected by Stau1 protein levels. Thus Stau1 may have a previously unrecognized role in the translational regulation of transcription-regulatory proteins” (Ricci, Kucukural, Cenik et al. 2014).
- Evidence indicates that “Staufen recognizes double-stranded RNA in a sequence-independent manner” — that is, binding of Staufen is primarily influenced by the secondary structure of the RNA (Ricci, Kucukural, Cenik et al. 2014).
- Stau1 can affect export of mRNAs from the nucleus to the cytoplasm either positively or negatively — though not greatly either way — with Stau1-binding to the 3' UTR having a positive effect, and binding to the coding region having a negative effect (Ricci, Kucukural, Cenik et al. 2014).
- “Higher Stau1 levels led to a preferential increase in ribosome density on high-GC-content mRNAs” (Ricci, Kucukural, Cenik et al. 2014).
-
Disordered protein as regulator of translation
“Intrinsically disordered proteins play important roles in cell
signalling, transcription, translation and cell cycle regulation.
Although they lack stable tertiary structure, many intrinsically
disordered proteins undergo disorder-to-order transitions upon binding to
partners. Similarly, several folded proteins use regulated
order-to-disorder transitions to mediate biological function”. And now
it’s been found that the phosphorylation of certain amino acids in an
intrinsically disordered protein can mediate the switch between its
translation-tolerant and translation-inhibiting role (Bah,Vernon,
Siddiqui et al. 2015, doi:10.1038/nature13999).
- The eukaryotic translation initiation factor 4E (eIF4E) binds to mRNA 5' caps in order to facilitate recruitment of the small ribosomal subunit to the mRNA. Subsequently, eIF4E is joined by other factors in a complex that enables initiation of translation. Certain proteins “play an important role in the regulation of translation” by binding to eIF4E and blocking the formation of the larger complex, thereby inhibiting translation. A recent study demonstrates that phosphorylation of eIF4E binding proteins “results in a disorder-to-order transition, bringing them from their binding-competent disordered state [when they could bind to eIF4E and inhibit translation] to a folded state incompatible with eIF4E binding”. “These results ... exemplify a new mode of biological regulation mediated by intrinsically disordered proteins” (Rhoades and Metskas 2015, doi:10.1016/j.tibs.2015.02.007; Bah, Vernon, Siddiqui et al. 2015, doi:10.1038/nature13999).
-
mRNA localization
- “mRNA localization often contributes to translational control. Reporting in Science, Moor et al. (2017) now show that many mRNAs and ribosomes are asymmetrically distributed along the apical-basal axis of enterocytes. Remarkably, when starved mice are fed, mRNAs encoding ribosomal proteins rapidly move to the ribosome-rich apical side to activate translation”. This research “documents that RNA localization can regulate global translation and dynamically respond to an external signal, namely, nutrient availability in a living organism” (Lasko 2017, doi:10.1016/j.devcel.2017.08.017).
-
“Meiotic progression is controlled by cytoplasmic polyadenylation and
translational activation of masked, maternal mRNAs. RNA-binding-protein
interactions with adjacent cis elements cause local conformational
changes to the mRNAs that determine the extent and timing of their
activation”. “Masked mRNAs have short poly(A) tails, whose extension can
be promoted by sequence-specific interactions between CPEB and the
cytoplasmic polyadenylation element (CPE), an RNA motif in the 3'
untranslated region (3' UTR). There are four CPEB proteins in
vertebrates. Phosphorylation of CPE-bound CPEB permits remodeling of the
RNP complex and promotes recruitment of a poly(A) polymerase that
elongates the tail of the mRNA. This in turn promotes recruitment of
translation initiation factors, thus activating translation”.
-
Targeting translation elongation
- “A PUF [Pumilio and Fem-3 binding factor]-Ago complex binds eukaryotic elongation factor 1A (eEF1A) and reduces its ability to hydrolyze GTP, an activity needed for delivery of aminoacylated tRNAs to translating ribosomes. The net result is attenuated translation elongation” and therefore reduced production of proteins (Pasquinelli 2012).
-
Alternative translation start sites
These should not be confused with alternative transcription start
sites, discussed under Alternative coding sequences
(transcription start and termination)
above.
- A recent study in “ribosomal profiling” was characterized in a Science article this way: "A startling feature [of the study] was the extent of the new translational start sites identified. Of the ~5000 genes examined, 13,454 likely start sites...were identified, with 65% of the mRNAs containing more than one start site and 16% with four or more” (Weiss and Atkins 2011). When the alternative start sites are “upstream” from the canonical one, additional protein sequence results; and when the alternative site is “downstream,” the protein is truncated.
-
Alternative translation termination
Ribosomes often cease translation when they encounter a stop codon — but
sometimes they don’t; instead they “read through” the stop codon, producing
carboxy-terminally extended nascent peptides that diversify the protein
collection of an organism. In eukaryotes, “readthrough is functionally
important insofar as it may suppress pathological phenotypes caused by
premature stop codons, antagonize nonsense-mediated decay and, by changing
the C-terminal sequence of a given protein, modulate its activity” (Dunn,
Foo, Belletier et al. 2013).
- Regarding a study of Drosophila: “Readthrough is far more pervasive than expected: the vast majority of readthrough events evolved within D. melanogaster and were not predicted phylogenetically. The resulting C-terminal protein extensions show evidence of selection, contain functional subcellular localization signals, and their readthrough is regulated, arguing for their importance. We further demonstrate that readthrough occurs in yeast and humans. Readthrough thus provides general mechanisms both to regulate gene expression and function, and to add plasticity to the proteome during evolution” (Dunn, Foo, Belletier et al. 2013).
- “Cotranslational degradation of polypeptide nascent chains plays a critical role in quality control of protein synthesis and the rescue of stalled ribosomes. In eukaryotes, ribosome stalling triggers release of 60S subunits with attached nascent polypeptides, which undergo ubiquitination by the E3 ligase Ltn1 and proteasomal degradation facilitated by the ATPase Cdc48 ... We examined how the canonical release factors Sup45-Sup35 (eRF1-eRF3) and their paralogs Dom34-Hbs1 affect the total population of ubiquitinated nascent chains associated with yeast ribosomes. We found that the availability of the functional release factor complex Sup45-Sup35 strongly influences the amount of ubiquitinated polypeptides associated with 60S ribosomal subunits, while Dom34-Hbs1 generate 60S-associated peptidyl-tRNAs that constitute a relatively minor fraction of Ltn1 substrates. These results uncover two separate pathways that target nascent polypeptides for Ltn1-Cdc48-mediated degradation and suggest that in addition to canonical termination on stop codons, eukaryotic release factors contribute to cotranslational protein quality control” (Shcherbik, Chernova, Chernoff and Pestov 2016, 10.1093/nar/gkw566).
-
Translational bypassing
For 25 years only a single case of “translational bypassing” has been
known (in a bacteriophage), whereby a section of an mRNA is ignored by
the ribosome during translation. “Bypassing requires translational
blockage at a ‘takeoff codon’ immediately upstream of a stop codon
followed by a hairpin, which causes peptidyl-tRNA dissociation and
reassociation with a matching ‘landing triplet’ 50 nucleotides downstream,
where translation resumes” (Lang, Jakubkova, Hegedusova et al. 2014). This
now looks set to change.
- “Here, we report 81 translational bypassing elements (byps) in mitochondria of the yeast Magnusiomyces capitatus and demonstrate in three cases, by transcript analysis and proteomics, that byps are retained in mitochondrial mRNAs but not translated”. The researchers report evidence that the bypassing elements are mobile genetic elements, capable of moving both within the same mitochondrial DNA and also between species. “Given the apparent mobility of byps and byp-like elements, it is conceivable that they also occur in mtDNAs outside fungi and in nuclear genomes” (Lang, Jakubkova, Hegedusova et al. 2014).
- rRNA modifications See “Ribosomal (rRNA and associated protein) modifications” under “RNA modifications” above.
-
Endoplasmic reticulum as regulator of translation
Recent findings, including those relating to the localization to the
endoplasmic reticulum (ER) of many factors that bind to and regulate mRNAs
and their translation, “suggest a biochemical and regulatory ER translation
environment that is distinct from the cytosol. Very recent reports also
reveal that the ER-associated translational system is dynamic, with the
capacity to rapidly reorganize in response to cellular stimuli or stress.
Taken together, these developments point to a need for re-examining our
understanding of how mRNA translation is spatially organized and regulated
in eukaryotic cells ... [There is] a newly emerging model for
mRNA translation on the ER, whereby the ER is a primary site of general
protein synthesis, as well as a site with exquisite regulatory functions
that can selectively influence specific mRNAs by several mechanisms. This
new model contributes to the ever-expanding richness of post-transcriptional
gene regulation and adds an important new variable of ER localization into
the consideration of how the translation of an mRNA may be regulated” (Reid
and Nicchitta 2015, doi:10.1038/nrm3958).
-
Stress granules and processing bodies as regulators of translation
Stress granules (SGs) and processing bodies (PBs) are non-membrane-enclosed
RNA granules that dynamically sequester translationally inactive messenger
ribonucleoprotein particles (mRNPs) into compartments that are distinct from
the surrounding cytoplasm. mRNP remodeling, silencing, and/or storage
involves the dynamic partitioning of closed-loop polyadenylated mRNPs into
SGs, or the sequestration of deadenylated, linear mRNPs into PBs. SGs form
when stress-activated pathways stall translation initiation but allow
elongation and termination to occur normally, resulting in a sudden excess
of mRNPs that are spatially condensed into discrete foci by protein:protein,
protein:RNA, and RNA:RNA interactions. In contrast, PBs can exist in the
absence of stress, when specific factors promote mRNA deadenylation,
condensation, and sequestration from the translational machinery. The
formation and dissolution of SGs and PBs reflect changes in messenger RNA
(mRNA) metabolism and allow cells to modulate the proteome and/or mediate
life or death decisions during changing environmental conditions”
(Ivanov, Kedersha and Anderson 2019, doi:10.1101/cshperspect.a032813).
-
Cytoskeleton as regulator of translation
See, for example Seyun Kim and Pierre A. Coulombe, “Emerging Role for the
Cytoskeleton as an Organizer and Regulator of Translation” in Nature
Reviews Molecular Cell Biology (2010): “Recent evidence favours the
hypothesis that the cytoskeleton participates in the spatial organization
and regulation of translation, at both the global and local level, in a
manner that is crucial for cellular growth, proliferation and function”.
-
Regulated “error” rates in protein synthesis
Accumulating evidence indicates that cells “regulate the synthesis of
mutant proteins molecules that deviate from the genetic code”. “For a long
time, it was commonly thought that translational errors must always be
avoided. Recent results indicate that cells actively regulate
translational errors for beneficial purposes. ... It is important to note
that reduced fidelity of replication and transcription have already been
shown to be beneficial in certain circumstances. For example, somatic
hypermutation reduces the fidelity of DNA replication by more than
1,000-fold and enables B cells to generate diverse libraries of receptors
and antibodies. The ~100-fold lower fidelity of retroviral reverse
transcriptase enables the generation of a diverse popultion of
retroviruses, some of which can better resist cellular and pharmacological
attacks. It should not be surprising that cells could have also evolved to
use regulated translational errors for stress response and adaptation” (Pan
2013).
- See also Co-translational mRNA decay under POST-TRANSCRIPTIONAL DECISION-MAKING —> RNA degradation.
-
Nuclear sequestration of mRNAs.
- “When under stress, cells switch to an energy-preserving, non-proliferative state, one hallmark of which is translation inhibition. Wang and colleagues now show that translation repression in these conditions involves the retention of polyadenylated mRNAs in the nucleus”. “SIRT1-mediated deacetylation of PABP1 inhibits translation by sequestering polyadenylated mRNAs in the nucleus. This mechanism seems to be part of an adaptive cellular response to energy deprivation that is integrated into cellular pathways regulating energy homeostasis. As nuclear retention of polyadenylated mRNAs has also been observed in response to stresses other than starvation, it is possible that suppression of nuclear export of polyadenylated mRNAs mediated by SIRT1–PABP1 (and/or other mechanisms) is more generally implicated in translation regulation in the event of stress” (Strzyz 2017, doi:10.1038/nrm.2017.82).
-
What hasn’t been said here. The foregoing is very cursory. Kong
and Lasko (2012) introduce a review of the subject (“Translational Control
in Cellular and Developmental Processes”) by indicating the range of
relevant topics: “we first discuss mechanisms that regulate the initiation
of translation by modulating the binding of essential translation
initiation factors to the 5' cap structure. Next, we review processes that
regulate translation by acting on the length of the poly(A) tail of target
mRNAs. In subsequent sections, we turn to the regulation of mRNAs that have
short open reading frames (ORFs) upstream of their main ORFs (these are
known as upstream ORFs (uORFs)) and then to translational control at later
stages of the process, such as ribosomal subunit joining and elongation.
Processes by which ribosomal proteins function outside ribosomes to
regulate translation is discussed in the next section, followed by a review
of how post-transcriptional modifications of nucleotides in ribosomal RNAs
(rRNAs) modulate ribosome function. Later sections of the Review include a
discussion of translational masking, the targeting of mRNAs into
translationally silent particles and how the phenomenon of mRNA
localization is coupled to translational regulation. Finally, examples are
given that demonstrate how these processes are related to human disease”.
The authors add that, “unlike transcriptional control, which is restricted to the nucleus, translational control mechanisms operate throughout the cell and can regulate expression of cytoplasmic proteins to ensure that they are present at the positions and times that they are required”. There is a powerful argument against genocentrism in all this.
The task of making a functional protein based on the “instructions” in a gene
is not finished after an mRNA has been translated into a protein. The protein
still needs to be (more or less) folded, and the way it is folded affects its
function. So, too, various chemical modifications to the protein can radically
alter its function. None of this later shaping of proteins can be said to be
directed by genes. Rather, it indicates how responsibility for specifying
protein extends far beyond DNA and the nucleus.
-
Histone and histone tail modifications
- (Note: these are post-translational in the sense that the histones and their tails are modified after the histone proteins have been produced. However, modifications to nucleosomal histones and their tails directly affect gene transcription, and are treated above under PRE-TRANSCRIPTIONAL DECISION-MAKING .)
-
Alternative protein folding
The results of all expression of protein-coding genes depend upon
the “correct” folding of the protein once it is translated. For any
typical protein, there is a vast number of theoretically possible
foldings, which the cell narrows down according to its own needs.
- Chaperone proteins help determine how a protein will fold, and therefore also how it will function.
- Alternative protein folding is a vast topic that basically remains untouched in this document.
-
Protein homeostasis network
- Regarding proteasomes, protein complexes that regulate protein amounts by degrading selected proteins (including damaged or no longer needed proteins): “The control of proteasome-mediated protein degradation is thought to occur mainly at the level of polyubiquitylation of the substrate. However, the proteasome can also be regulated directly, as now demonstrated by a study in which DYRK2-mediated phosphorylation of the 19S subunit Rpt3 [of the proteasome] is found to increase proteasome activity” (blurb for Huibregtse and Matouschek 2016, doi:10.1038/ncb3306).
- Genes are employed in the production of proteins, but the net rate of accumulation for any protein depends on the rates of mRNA and protein degradation and the rate of cell division (with its dilution of proteins), as well as the rate of gene transcription. (For coverage of one of these topics, see RNA degradation above.) Protein half-lives have not been well studied as yet, but in cells growing at a moderate rate they may commonly range from 1 hour to 1 day (Plotkin 2011). There are various pathways of degradation, requiring careful regulation.
- More generally, there is an elaborate molecular network responsible for keeping the overall complement of proteins in a cell or organism healthy and in balance. This protein homeostasis (or proteostasis) network, which varies considerably between different cell types, has two complementary functions: a protein folding function and a protein degradation function. “The network has the potential to provide global management to overcome loss- and/or gain-of-function mutations associated with numerous protein misfolding diseases. This conclusion is consistent with the fact that the standard can be varied between cell types and even with a given cell to meet folding demands in response to a broad range of signaling pathways, including the unfolded protein response, heat shock response, oxidative stress response, diet restriction, inflammatory signaling, and insulin growth factor 1 receptor signaling, all of which help protect the cell from environmental insults and during aging” (Hutt and Balch 2010).
- “A protein degradation pathway is found at the inner nuclear membrane that is distinct from, but complementary to, endoplasmic-reticulum-associated protein degradation, and which is mediated by the Asi protein complex; a genome-wide library screening of yeast identifies more than 20 substrates of this pathway, which is shown to target mislocalized integral membrane proteins for degradation” (blurb in Nature for doi:10.1038/nature14096).
- “While cellular proteins were initially thought to be stable, research over the last decades has firmly established that intracellular protein degradation is an active and highly regulated process: Lysosomal, proteasomal, and mitochondrial degradation systems were identified and found to be involved in a staggering number of biological functions. Here, we provide a global overview of the diverse roles of cellular protein degradation using seven categories: homeostasis, regulation, quality control, stoichiometry control, proteome remodeling, immune surveillance, and baseline turnover” (McShane and Selbach 2022, doi:10.1146/annurev-cellbio-120420-091943).
-
Post-translational modification of regulatory proteins
This section is a tiny fragment relative to the vast content rightly
belonging to the topic of post-translational modification of proteins.
Countless proteins figuring in virtually all aspects of gene expression
are regulated by PTMs, often in complex, spatially and temporally tuned
ways. A few relevant examples are mentioned throughout this document.
(Searching on “post-translational modification” and “PTM” will turn up
many of them.)
“Post-translational modifications, which are found largely in intrinsically
disordered protein regions, regulate protein activity, stability and
interactions with partners. They are therefore critical for controlling
essentially all cellular processes. A single modification event can have
dramatic effects; however, proteins are often modified on multiple sites to
collectively modulate the biological outcome. Multiple PTMs can mediate the
same, complementary or opposing effects and the result of their interplay is
determined by a complex combination of the number, positioning and type of
modifications. Multiple PTMs can also synergize to shift the conformational
or binding equilibria of the modified protein to modulate its interaction
with partners or formation of higher order assembly. Recognition of such PTM
crosstalk is crucial for understanding the underlying mechanisms of complex
regulatory processes”
(Csizmok and Forman-Kay 2018, doi:10.1016/j.sbi.2017.10.013).
Once a protein is synthesized and folded properly, it is subject to all
manner of chemical modifications in connection with the ever-changing
metabolism of cell and organism. Here we deal mainly with the
modification of those particular proteins (excluding histones,
treated above) that are considered to be more or less direct regulators
of gene expression. But it is worth keeping in mind that all
functional modification of proteins affects gene expression, inasmuch as
genes are commonly taken to be the determiners of specific proteins.
For a protein with one or more PTMs commonly becomes, in functional terms, a
different protein — and sometimes a radically different one.
“Metabolites affect cell growth in two different ways. First, they serve as
building blocks for biomass accumulation. Second, metabolites regulate the
activity of growth-relevant signaling pathways. They do so in part by
covalently attaching to proteins, thereby generating post-translational
modifications (PTMs) that affect protein function, the focus of this
Perspective. Recent advances in mass spectrometry have revealed a wide
variety of such metabolites, including lipids, amino acids, Coenzyme-A,
acetate, malonate, and lactate to name a few. An active area of research is
to understand which modifications affect protein function and how they do
so. In many cases, the cellular levels of these metabolites affect the
stoichiometry of the corresponding PTMs, providing a direct link between
cell metabolism and the control of cell signaling, transcription, and cell
growth” (Figlia, Willnow and Teleman 2020, doi:10.1016/j.devcel.2020.06.036).
(Relative to previous item:)
“Upon oxidative stress, glutathionylation of NFκB decreases its ability to
interact with DNA, 2001), thus aligning gene expression with changes in
redox state. Analogously, modification of histones with the negatively
charged glutaryl and succinyl groups causes nucleosome destabilization,
thus allowing increased gene transcription”
(Figlia, Willnow and Teleman 2020, doi:10.1016/j.devcel.2020.06.036).
- Methylation of arginine residues. “Protein methylation [of arginine residues] of coactivators, transcription factors, and signal transducers, among other proteins, plays important roles in transcriptional regulation. Protein methylation may affect protein-protein interaction, protein-DNA or protein-RNA interaction, protein stability, subcellular localization, or enzymatic activity. Thus, protein arginine methylation is critical for regulation of transcription” (Lee and Stallcup 2010). Arginine methyltransferases, which apply methyl groups to arginine residues, are themselves transcription co-activators.
-
Phosphorylation. The phosphorylation, particularly of serine
residues, is the most widely studied post-translational modification of
proteins. Among many other things, it plays a huge role in the
regulation of signaling pathways that in turn regulate gene expression.
To provide a hint of the regulatory complexities involved, here is one
barely sketched example relating to the tumor suppressor protein (a
transcription factor) known as “p53":
- p53 is normally inactivated by a negative regulator, a protein called “MDM2,” but becomes active when the cell experiences certain kinds of damage or stress. Then enzymes phosphorylate one end of p53, as a result of which other proteins are recruited, and the shape of p53 is changed. All this leads to its dissociation from the repressive MDM2. The changes also allow the binding of transcriptional co-activators, which then acetylate the opposite end of p53, thereby exposing the protein sequence that binds to DNA and activates or represses specific genes — all in the interest of bringing about the death of the cell if its damage is irreparable, or its repair otherwise. In the event of successful repair, other enzymes deacetylate p53 so that it does not cause cell death. In this way, p53 can help to prevent cancer. (Adapted from the Wikipedia entry, “p53".)
- Neurogenesis is initiated by the transient expression of certain proneural proteins (transcription factors). “Phosphorylation of a single Serine at the same position in Scute and Atonal proneural proteins governs the transition from active to inactive forms by regulating DNA binding. The equivalent Neurogenin2 Threonine also regulates DNA binding and proneural activity in the developing mammalian neocortex. Using genome editing in \*IDrosophila\*i, we show that Atonal outlives its mRNA but is inactivated by phosphorylation. Inhibiting the phosphorylation of the conserved proneural Serine causes quantitative changes in expression dynamics and target gene expression resulting in neuronal number and fate defects. Strikingly, even a subtle change from Serine to Threonine appears to shift the duration of Atonal activity in vivo, resulting in neuronal fate defects” (Quan, Yuan, Tiberi et al. 2016, doi:10.1016/j.cell.2015.12.048).
- “Many tissues harbor a reservoir of stem cells that remains quiescent but can be activated as needed for growth and repair. How cells enter, maintain, and then exit quiescence is incompletely defined. Studying skeletal muscle stem cells in mice, Zismanov et al. reveal a role for translational repression. Stem cell quiescence requires phosphorylation (a posttranslational protein modification) of the translation initiation factor eIF2α at a particular amino acid residue; dephosphorylation (removal of the phosphoryl group) or blocking phosphorylation causes muscle stem cells to exit quiescence and differentiate. Moreover, inhibiting dephosphorylation leads muscle stem cells to self-renew and regenerate” (Purnell 2016 [Science vol. 351, p. 377], reporting on an article in Cell Stem Cell vol. 18, p. 79.).
- From an article entitled, “Tob2 Phosphorylation Regulates Global mRNA Turnover to Reshape Transcriptome and Impact Cell Proliferation”: “Tob2, an anti-proliferative protein, promotes deadenylation through recruiting Caf1 deadenylase to the mRNA poly(A) tail by simultaneously interacting with both Caf1 and poly(A)-binding protein (PABP). Previously, we found that changes in Tob2 phosphorylation can alter its PABP-binding ability and deadenylation-promoting function ... In this study, we found that c-Jun amino-terminal kinase (JNK) increases phosphorylation of Tob2 at many Ser/Thr sites in the intrinsically disordered region (IDR) that contains two separate PABP-interacting PAM2 motifs. JNK-induced phosphorylation or phosphomimetic mutations at these sites weaken the Tob2–PABP interaction. In contrast, JNK-independent phosphorylation of Tob2 at serine 254 (S254) greatly enhances Tob2 interaction with PABP and its ability to promote deadenylation ... Our findings reveal a novel mechanism by which Ccr4–Not complex is recruited by Tob2 to the mRNA 3′ poly(A)-PABP complex in a phosphorylation dependent manner to promote rapid deadenylation and decay across the transcriptome, eliciting transcriptome reprogramming and suppressed cell proliferation” (Chen, Strouz, Huang1 and Shyu 2020, doi:10.1261/rna.073528.119).
-
Sumoylation
“Sumoylation is a reversible modification where the ubiquitin-like
protein Sumo is attached to one or more lysine residues in a target
protein. Thousands of proteins are Sumo targets. Cell stress can induce
sumoylation of many proteins, an effect often referred to as the Sumo
stress response. Transcription factors (TFs) and chromatin modifiers are
among the most prominent Sumo substrates, and although recent studies in
budding yeast and mammalian cells have shown that Sumo can activate
transcription, sumoylation of TFs in response to cell stress is generally
associated with inhibition of transcription”
(Enserink 2017, doi:10.1002/bies.201700065).
“SUMO is an essential modification that helps cells and organisms to cope
with stress. It modulates many cellular processes, but the majority of
its functions are linked to nuclear activities. There is increasing
evidence that SUMOylation can function as both a precise and a
promiscuous modifier, with different roles in different cellular
processes. Active participation of SUMO machinery components as
coregulators of transcription is emerging as one way to regulate SUMO
targets spatially. Group SUMOylation of chromatin seems to be important
for SSR as well as for the regulation of transcription at promoters and
enhancers. Importantly, there is convincing evidence suggesting that the
regulation of transcription by SUMOylation includes modulation of Pol2
[RNA polymerase II] pausing in stress. SUMOylation might also be
important for the maintenance and organisation of chromatin structure at
enhancer-promoter contacts and especially at chromatin anchors that
define the TAD boundaries ... the mechanisms underpinning the SUMO stress
response in the regulation of chromatin-linked processes are only
beginning to emerge and will keep us busy learning more about this
fascinating protein modification”
(Niskanen and Palvimo 2017, doi:10.1002/bies.201600263).
- SUMO [Small Ubiquitin-like-Modifier] can “elicit diverse downstream consequences following conjugation to different proteins”. The specific effects are likely determined in part “by the intersection [crosstalk] with other posttranslational modification pathways, including ubiquitylation, phosphorylation, and acetylation” (Cubeñas-Potts and Matunis 2013).
- “We provide proteomic evidence for sumoylation of 3,617 proteins at 7,327 sumoylation sites, and insight into SUMO group modification by clustering the sumoylated proteins into functional networks. The data support sumoylation being a frequent protein modification (on par with other major protein modifications) with multiple nuclear functions, including in transcription, mRNA processing, DNA replication and the DNA-damage response” (Hendriks and Vertegaal 2016, doi:10.1038/nrm.2016.81).
- “Stress-induced changes in sumoylation are the result of an altered balance between the activity of enzymes that carry out sumoylation and the desumoylating enzymes. However, what is less clear is how SSR [Sumo stress response] specificity is achieved. For instance, DNA damage triggers sumoylation of multiple proteins involved in DNA repair, but other stresses, such as nutrient stress, do not affect sumoylation of these very same proteins. Furthermore ... sumoylation of some groups of transcription factors and chromatin remodelers increases during cell stress, whereas others become desumoylated. Clearly, how SSR specificity is achieved is still poorly understood” (Enserink 2017, doi:10.1002/bies.201700065).
- For more on sumoylation, see “Sumoylation” under “Histone tail modifications above.
-
Ubiquitination
“Nascent proteins are at risk of misfolding, ubiquitination, and
proteasomal degradation. However, ubiquitination does not always signify
the end for proteins. We propose a model that uses ubiquitination as a
“fix me” signal for misfolded nascent proteins. Ubiquitination may
recruit unfolding enzymes, deubiquitinases, and chaperones to provide
additional chances for aberrant nascent proteins to fold properly”
(Culver, Li and Mariappan 2022, doi:10.1002/bies.202200014).
-
Glycosylation and O-GlcNAcylation
“Post-translational modifications (PTMs) immensely expand the diversity
of the proteome. Glycosylation, among the most ubiquitous PTMs, is a
dynamic and multifarious modification of proteins and lipids that
generates an omnipresent foliage on the cell surface. The resulting
protein glycoconjugates can serve important functions in biology.
However, their vast complexity complicates the study of their structures,
interactions, and functions ... In this review, we discuss the growing
forestry toolbox to characterize the structure, interactions, and
biological functions of protein glycoconjugates”.
“There is an emerging realization that glycoconjugates are more than the
sum of their individual glycan and protein components and that
understanding their biology at the conjugate level can pay enormous
dividends in biomarker and drug discovery”
(Critcher, Hassan and Huang 2022, doi:10.1016/j.tibs.2022.02.007).
O-GlcNAcylation is “the ubiquitous, dynamic, and reversible
addition of a sugar motif (β-D-N-acetylglucosamine) to serine and
threonine residues”. “O-GlcNAcylation helps regulate gene
expression by (1) changing the properties of transcription factors
(localization, stability, DNA binding, and transcriptional activity); (2)
directly or indirectly modifying histones; (3) impacting DNA methylation
through modulation of DNA methyltransferase 1 (DNMT1) and ten–eleven
translocation 1, 2 and 3 (TET1, 2 and 3) protein properties (activity for
DNMT; stability and DNA binding for TET); and (4) regulating RNA
polymerase II transcription at the initiation and elongation stages.
Moreover, OGT [an enzyme that adds the GlcNAc moiety to proteins]
interacts with and regulates proteins in polycomb repressive complexes
(PRCs) 1 and 2, and a recent study reported that O-GlcNAcylation
levels contribute to the intron retention process. Finally, as evidence
of its broad impact on gene expression, O-GlcNAcylation dictates
the translational regulation of mRNAs modified with
N6-methyladenosine (m6A) through YTH
domain-containing m6A-RNA-binding proteins. Recently developed
approaches have enabled considerable progress in identifying
O-GlcNAcylated proteins and in unraveling the role of
O-GlcNAcylation in numerous biological processes. To date, the set
of O-GlcNAcylated proteins in humans, known as the
O-GlcNAcylome, consists of 8000 proteins and continues to grow”
(Dupas, Lauzier and McGraw 2023, doi:10.1186/s13072-023-00523-5).
Originally classified as post-transcriptional regulators of gene expression,
noncoding RNAs of various sorts are now known to be involved in gene regulation
at more than one level. Noncoding RNAs “contain extensive information stored
in the form of specific structural conformation or nucleotide sequence that
goes beyond the genetic code used for translation of protein-coding genes”
(Ørom and Shiekhattar 2011).
Regarding small RNAs in general: “We found that >90% of the small RNAs present
in the early X. tropicalis [frog] embryo could not be identified by
comparison with known small RNAs. ... This suggested that there are many small
RNAs and possible regulatory mechanisms in the early embryo that we do not yet
understand” (Harding, Horswell, Heliot et al. 2014). And again: large numbers
of small regulatory RNAs are invisible to prevailing approaches for identifying
them. This limits “the otherwise vast world of rsRNAs [regulatory small RNAs]
mainly to hair-pin loop bred typical miRNAs. The present study has analyzed for
the first time a huge volume of sequencing data from 4997 individuals and 25
cancer types to report 11,234 potentially regulatory small RNAs which appear to
have deep reaching impact ... Several of the potential rsRNAs have emerged as a
critical cancer biomarker ... The possible degree of cell system regulation by
sRNAs appears to be much higher than previously assumed” (Jha, Panzade, Pandey
and Shankar 2015, doi:10.1093/nar/gkv871).
“In general, sncRNAs [structured noncoding RNAs] forming RNPs
[ribonucleoproteins] are hundreds to thousands of times more abundant than
their mRNA counterparts. Surprisingly, only 50 sncRNA genes produce half of
the non-rRNA transcripts [longer than 60 nucleotides] detected in two different
cell lines. Together the results indicate that the human transcriptome is
dominated by a small number of highly expressed sncRNAs specializing in
functions related to translation and splicing”. “The transcriptome of model
cell lines is defined by a small number of highly expressed noncoding genes and
a large number of moderately expressed protein-coding genes”.
“Ribonucleoprotein particles are generated from highly abundant noncoding RNA
and proteins produced by uniformly less abundant protein-coding RNA”.
(Boivin, Deschamps-Francoeur, Couture et al. 2018, doi:10.1261/rna.064493)
“Often the noncoding genome’s functions are carried out by their RNA
transcripts, which may rely on their structures and/or extensive interactions
with other molecules ... [New technologies have] revealed surprising
versatility of RNA to participate in diverse molecular systems. For example,
tens of thousands of RNA–RNA interactions have been revealed in cultured cells
as well as in mouse brain, including interactions between transposon-produced
transcripts and mRNAs. In addition, most transcription start sites in the human
genome are associated with noncoding RNA transcribed from other genomic loci.
These recent discoveries expanded our understanding of RNAs’ roles in chromatin
organization, gene regulation, and intracellular signaling”
(Nguyen, Zaleta-Rivera, Huang et al., doi://10.1016/j.tig.2018.08.001).
-
Noncoding RNA in general
- Noncoding RNAs are being found to be involved in very many of the other aspects of gene regulation discussed in this document. For example: by virtue of their ability to complement DNA sequences, they can provide a means for targeting proteins such as chromatin remodeling proteins to particular locations in the genome. “Studies suggest a general role in gene regulation where ncRNAs can mark and often modulate the active chromatin state in both positive and negative manners” (Flynn and Chang 2012).
- Likewise, “enhancer elements and promoters are dispersed throughout the genome, and yet histone methyltransferases...and histone demethylases...are able to localize to these specific regions and in a cell-type specific manner, targeting their enzymatic function. ... Observations suggest that RNA can provide a gene-specific targeting mechanism to non-specific enzymatic activity” (Flynn and Chang 2012).
- A paper on small RNAs in bacteria hints at some of the basis for the kind of complexity illustrated in the sections below: the Qrr3 small RNA “uses four distinct mechanisms to control its particular targets: the Qrr3 sRNA represses luxR through catalytic degradation, represses luxM through coupled degradation, represses luxO through sequestration, and activates aphA by revealing the ribosome binding site while the sRNA itself is degraded. Qrr3 forms different base-pairing interactions with each mRNA target, and the particular pairing strategy determines which regulatory mechanism occurs ... the specific Qrr regulatory mechanism employed governs the potency, dynamics, and competition of target mRNA regulation” (Feng, Rutherford, Papenfort et al. 2015, doi:10.1016/j.cell.2014.11.051).
- Regarding small RNAs (small interfering RNAs, microRNAs, and piRNAs): the molecules involved in configuring these RNAs and assembling them into working complexes “are themselves regulated, thus providing additional layers to [transcriptional] silencing control” (Ipsara and Joshua-Tor 2015, doi:10.1038/nsmb.2931).
- “A paradigm is emerging in human cells, which proposes that non-coding RNAs, both small and long forms, function through the action of [the DNA methylating enzyme] DNMT3a to modulate chromatin and epigenetic states of gene expression [at genomic sites targeted by the non-coding RNAs]. While there are several other mechanisms of action described for lncRNAs in human cells, the interactions with DNMT3a and targeting of transcriptional and epigenetic states is of particular interest, as this mode of gene regulation has the potential to be long-lasting, heritable and may be of significant relevance to the development of targeted therapeutics” (Weinberg and Morris 2016, doi:10.1093/nar/gkw139).
- “The DNA in regions upstream of the transcription start site (TSS) of active genes must be accessible to the transcription apparatus. To allow this, promoters are characterized by a region that is free of nucleosomes: the nucleosome-depleted region (NDR). Classical representations depict NDRs with a sharp, unidirectional TSS. However, RNA polymerase (RNAP) II transcription from NDRs is inherently bidirectional. In some cases, this drives divergent gene expression, but more commonly the products are an mRNA plus an unstable, non-protein-coding RNA (ncRNA) generated on the other strand. The termination regions of protein-coding genes are also permissive for the synthesis of antisense-oriented ncRNAs, possibly due to an open chromatin structure here. Moreover, transcription is subject to substantial stochastic noise, with low-level RNAPII transcription, detectable over almost the entire genome. Together, these mechanisms generate very large numbers of ncRNAs. These ncRNAs are potentially damaging but are constantly cleared by the RNA surveillance machinery. Degradation is generally through 5′ or 3′ exonucleases, particularly the RNA exosome. In yeast, the Nrd1/Nab3/Sen1 (NNS) complex recognizes short consensus motifs in RNA sequences and triggers both ncRNA transcription termination and exosome recruitment to degrade the product” (Turowski and Tollervey 2020, doi:10.1016/j.tig.2020.05.011). But see also next item.
- In Saccharomyces cerevisiae (S. cerevisiae): “By using a noncoding transcription-inducible strain, we analyze the relationship between antisense elongation and coding sense repression, nucleosome occupancy, and transcription-associated histone modifications using near-base pair resolution techniques. We show that antisense noncoding transcription leads to the deacetylation of a subpopulation of −1/+1 nucleosomes associated with increased H3K36me3. Reduced acetylation results in the decreased binding of the RSC chromatin remodeler at −1/+1 nucleosomes and subsequent sliding into the nucleosome-depleted region hindering pre-initiation complex association. Finally, we extend our model by showing that natural antisense noncoding transcription significantly represses ∼20% of S. cerevisiae genes through this chromatin-based transcription interference mechanism” (Gill, Mafioletti, García-Molinaro and Stutz 2020, doi:10.1016/j.celrep.2020.107612).
- “As we continue to find new regulatory roles for RNAs, a theme is emerging in which regulation may not be mediated through the actions of a specific RNA, as one typically thinks of a regulator and target, but rather through the collective nature of many RNAs, each contributing a small degree of the regulatory load. This mechanism has been termed “crowd-control” and may apply broadly to miRNAs and to RNAs that bind and regulate protein activity. This provides an alternative way of thinking about how RNAs can act as biological regulators and has repercussions, both for the understanding of biological systems, and for the interpretation of results in which individual members of the “crowd” can replicate the effects of the crowd when overexpressed, but are not individually significant biological regulators” (Bracken 2023; doi:10.1261/rna.079644.123).
-
MicroRNA (mirna) activity
MicroRNAs (miRNAs) are a set of small (approximately 22 nucleotides)
non-protein-coding RNAs that regulate gene expression, especially at the
post-transcriptional level. The wide-ranging processes by which they are
formed, modified, and exert their influence — all in temporally and
spatially significant patterns — make them one of the most fundamental
regulatory elements of the organism. “Each microRNA may repress up to
hundreds of transcripts, and thus, it is estimated that microRNAs regulate
a large proportion of the transcriptome” (Salmena, Poliseno, Tay et al.
2011).
“Hundreds of microRNAs (miRNAs) are expressed in distinct spatial and
temporal patterns during embryonic and postnatal mouse development. The loss
of all miRNAs through the deletion of critical miRNA biogenesis factors
results in early lethality. The function of each miRNA stems from their
cumulative negative regulation of multiple mRNA targets expressed in a
particular cell type. During development, miRNAs often coordinate the timing
and direction of cell fate transitions. In adults, miRNAs frequently
contribute to organismal fitness through homeostatic roles in physiology”
(DeVeale, Swindlehurst-Chan and Blelloch 2021,
doi:10.1038/s41576-020-00309-5).
“miRNAs play a central role in establishing the spatiotemporal gene
expression patterns required to establish specialized cell types and
promote developmental complexity. The inherent complexity of miRNA
function, however, requires a scientific approach in which context-specific
miRNA function must be acknowledged”. “The expression pattern of a
specific miRNA may see it predominantly expressed at a particular stage of
development, enriched within an individual cell type, and localized to a
specific subcellular compartment” (Carroll, Tooney and Cairns 2013).
“Owing to their ability to simultaneously silence hundreds of target genes,
[miRNAs] have key roles in large-scale transcriptomic changes that occur
during cell fate transitions. In somatic stem and progenitor cells — such
as those involved in myogenesis, haematopoiesis, skin and neural
development — miRNA function is carefully regulated to promote and
stabilize cell fate choice. miRNAs are integrated within networks that
form both positive and negative feedback loops. Their function is regulated
at multiple levels, including transcription, biogenesis, stability,
availability and/or number of target sites, as well as their cooperation
with other miRNAs and RNA-binding proteins. Together, these regulatory
mechanisms result in a refined molecular response that enables proper
cellular differentiation and function” (Shenoy and Blelloch 2014).
“Specifically, it has been reported that miRNAs establish thresholds in the
response of their targets to transcriptional induction, reduce the
cell-to-cell variability of target gene expression and induce correlations
between the expression of various targets within individual cells. Which of
these mechanisms is relevant in a particular context is an essential yet
difficult question to answer because the underlying interaction networks
are large, complex and only partially known. Thus, much of the current
debate in the field oscillates between defining (and redefining) what a
miRNA target is and determining the appropriate readout of miRNA–target
interactions, while taking into account that the impact of a miRNA on
individual targets depends on many dynamic factors. Among these factors are
the cellular localization of miRNAs and their targets, their relative
concentrations and the context-specific effects of other regulators,
including transcription factors and RNA-binding proteins” (Hausser and
Zavolan 2014, doi:10.1038/nrg3765).
“Our comprehensive and highly consistent data set from several
high-throughput technologies provides strong evidence that
context-dependent microRNA target sites (CDTS) are as frequent and
functionally relevant as constitutive target sites. Furthermore, we found
the global context to be insufficient to explain the CDTS, and that
flanking sequence motifs provide individual context that is an equally
important factor. Our results demonstrate that, similar to TF-mediated
regulation, global and individual context dependency are prevalent in
microRNA-mediated gene regulation, implying a much more complex
post-transcriptional regulatory network than is currently known” (Veloso,
Kirkconnell, Magnuson et al. 2014, doi:10.1101/gr.171405.113).
“Another factor to consider is that the regulatory network of a miRNA is
probably dynamic. As each individual cell expresses only a subset of genes
and transcript isoforms, only a proportion of the miRNA complementary sites
that are annotated transcriptome wide will be present and relevant in any
given cell. Furthermore, the various miRNA-binding sites are likely to
differ in their affinity for the miRNA-loaded AGO protein, and different
sites are likely to be saturated at different concentrations of miRISC
[miRNA-induced silencing complex]. Finally, RNA-binding proteins modulate
the accessibility of individual sites in a tissue-specific manner” (Hausser
and Zavolan 2014, doi:10.1038/nrg3765).
“miRNAs can regulate a high number of target mRNAs; for instance, a single
miRNA can affect the expression of over 100 transcripts and, conversely, a
given mRNA can contain target sites for a large number of miRNAs, which
suggests a complex regulatory network whose logic remains largely
unexplored” (Guil and Esteller, doi:10.1016/j.tibs.2015.03.001).
The miRNA story is one of ever-increasing complexity. Two researchers
report “a surprising fragmentation in the miRISC functional pool, striking
differences in the availability of miRNA families and saturability of
miRNA-mediated silencing. Furthermore, we provide direct experimental
evidence that only a limited subset of miRNAs, defined by a conjuncture of
expression threshold, miRISC availability and low target site abundance, is
susceptible to competitive effects through microRNA-binding sites”. “We
postulate that the different scenarios of expression, availability and
stoichiometry experimentally revealed here can be selected to serve distinct
physiological purposes and may unlock different properties of the miRISC
machinery. Extreme abundance of a simple miRISC pool, programmed by a single
miRNA family, such as miR-430 in the zebrafish embryo, is a logical fit for
the rapid clearance of maternal transcripts at Maternal-to-Zygotic
Transition (MZT). A different scenario should prevail in fully
differentiated somatic cells reaching a homeostatic state. In this case,
modest to moderate changes in expression of miRNAs, several of which may act
redundantly, will rarely result in a drastic phenotype. Nonetheless, a
subset of miRNAs should lie in the dynamic or responsive range of
concentrations, with the available pool of miRISC being near-stoichiometric
with biologically critical mRNA targets to allow a sensitive modulation of
silencing in response to environmental and signalling cues. Re-visiting the
properties of the miRISC in each of these states will potentially resolve
yet more complexity in miRNA-mediated silencing mechanisms” (Mayya and
Duchaine 2015, doi:10.1093/nar/gkv720).
“MicroRNAs often occur in families whose members share an identical 5′
terminal ‘seed’ sequence. The seed is a major determinant of miRNA activity,
and family members are thought to act redundantly on target mRNAs with
perfect seed matches, i.e. sequences complementary to the seed. However,
recently sequences outside the seed were reported to promote silencing by
individual miRNA family members ... Using the let-7 miRNA family in
Caenorhabditis elegans, we find that seed match imperfections can
increase specificity by requiring extensive pairing outside the miRNA seed
region for efficient silencing and that such specificity is needed for
faithful worm development. In addition, for some target site architectures,
elevated miRNA levels can compensate for a lack of complementarity outside
the seed. Thus, some target sites require higher miRNA concentration for
silencing than others, contrasting with a traditional binary distinction
between functional and non-functional sites. We conclude that changing
miRNA concentrations can alter cellular miRNA target repertoires. This
diversifies possible biological outcomes of miRNA-mediated gene regulation
and stresses the importance of target validation under physiological
conditions to understand miRNA functions in vivo”
(Brancati and Großhans 2018, doi:10.1093/nar/gky201).
“The findings by Dallaire et al. [on Caenorhabditis elegans] strongly
suggest that miRISC binding does not by itself destine a transcript for
degradation. The fate of a transcript is highly dependent on the composition
and subcellular localization of miRISC. Tissue specificity and temporal
specificity of expression, as well as modifications to miRNAs, target mRNAs,
and miRISC components all contribute to the rich dynamics of
miRNA-mediated post-transcriptional gene regulation during development and
beyond” (Galagali and Kim 2018, doi:10.1016/j.devcel.2018.10.009).
“Animal germ cells possess a specific post-transcriptional regulatory
context allowing the storage of maternal transcripts in the oocyte until
their translation at a specific point in early development. As key
regulators of gene expression, miRNAs repress translation mainly through
mRNA destabilization. Thus, germline miRNAs likely use distinct ways to
regulate their targets. Here, we use C. elegans to compare miRNA
function within germline and somatic tissues. We show that the same miRNA
displays tissue-specific gene regulatory mechanisms. While translational
repression occurs in both tissues, targeted mRNAs are instead stabilized in
the germline. Comparative analyses of miRNA silencing complexes (miRISC)
demonstrate that their composition differs from germline to soma. We show
that germline miRNA targets preferentially localize to perinuclear regions
adjacent to P granules, and their repression is dependent on the core P
granule component GLH-1. Together, our findings reveal the existence of
different miRISC in animals that affect targeted mRNAs distinctively”
(Dallaire, Frédérick, and Simard 2018, doi:10.1016/j.devcel.2018.08.022).
“Several layers of tightly controlled regulation have evolved to maintain
the levels of mature miRNAs in order to fine-tune gene expression during
development and differentiation. Such multifaceted regulation ultimately
prevents gross changes in gene expression that can contribute to numerous
diseases. Among these mechanisms of control, post-transcriptional steps are
predominant, and increasing evidence shows the central role of general RBPs
[RNA-binding proteins] in the control of miRNA production. The binding of
RBPs to TL [terminal loop] sequences within miRNA precursors (pri- and
pre-miRNAs) has emerged as a general mechanism to regulate the activity of
DROSHA and/or DICER. This can encompass different mechanisms, such as
conformational changes and dynamic destabilization induced by the binding of
these auxiliary factors. For example, the binding of hnRNP A1 or Rbfox
proteins to pri-miRNAs leads to structural changes that affect
Microprocessor binding and/or activity. Another common mechanism is
antagonistic binding of the regulatory RBP to either a positive or negative
regulator, as seen with the competitive binding of hnRNP A1 and KSRP to
let-7 precursors in differentiated cells, or MBNL-1 antagonizing LIN28
binding to pri-mir-1”
(Michlewski and Cáceres 2018, doi:10.1261/rna.068692.118).
“While canonical miRNA targeting involves pairing of the miRNA seed,
nucleotides 2–7 of the miRNA, to target 3′ UTR sequences, recent studies
have revealed roles for miRNA sequences beyond this region in specifying
target recognition and regulation.
Auxiliary base pairing to sequences in the 3′ half of the miRNA can overcome
seed imperfections and confer specificity for individual members of a miRNA
family that share identical seed sequences.
Base pairing of 3′-end miRNA sequences enables targeting of protein-coding
sequences that lack canonical seed-pairing interactions.” Further, “the
extent of pairing to the miRNA 3′ end can influence the stability of the
miRNA itself” — that is, “extensive pairing interactions between a miRNA and
its target can lead to target-directed miRNA degradation”
(Chipman and Pasquinelli 2019, doi:10.1016/j.tig.2018.12.005).
“The mammalian neocortex has six layers of distinct neurons generated
sequentially during embryogenesis by stem cells that change competence in
producing specific neuronal types as neurogenesis progresses. Shu et al.
show miR-128, miR-9, and let-7 form temporally opposite and functionally
antagonistic expression/activity gradients to time changes in stem-cell
competence” (TOC blurb for
Shu, Wu, Ruan et al. 2019, doi:10.1016/j.devcel.2019.04.017).
“Neuroscientists are keen to tap into the brain's reservoir of neural stem
cells to treat age-associated cognitive decline, neurodegeneration, and
other illnesses. In the mouse brain, quiescent stem cells in the central
subependymal zone are poised to produce new neuroblasts during adulthood.
Lepko et al. have linked mobile microRNA signals to regulation of stem cell
quiescence. Low amounts of neurogenic fate determinants can be expressed in
the stem cells as RNA but are not translated into proteins. Analysis of
posttranscriptional regulation fingered the microRNA miR-204, which is
abundant in the brain, as the block on the stem cells. The source of miR-204
was the choroid plexus, which delivered miR-204 into the cerebrospinal fluid
and, from there, to the neural stem cells. Thus, at the blood–brain
interface, the choroid plexus is ready to integrate external, systemic
signals with demands for adult brain neurogenesis”
(blurb in Science for a 2019 article in Embo Journal:
doi:10.15252/embj.2018100481).
“MicroRNAs (miRNAs) play roles in diverse developmental and disease
processes. Distinct miRNAs have hundreds to thousands of conserved mRNA
binding sites but typically direct only modest repression via single sites.
Cotargeting of individual mRNAs by different miRNAs could potentially
achieve stronger and more complex patterns of repression. By comparing
target sets of different miRNAs, we identified hundreds of pairs of miRNAs
that share more mRNA targets than expected (often by twofold or more)
relative to stringent controls. Genetic perturbations revealed a functional
overlap in neuronal differentiation for the cotargeting pair
miR-138/miR-137. Clustering of all cotargeting pairs revealed a group of
nine predominantly brain-enriched miRNAs that share many targets. In
reporter assays, subsets of these miRNAs together repressed gene expression
by five- to 10-fold, often showing cooperative repression. Together, our
results uncover an unexpected pattern in which combinations of miRNAs
collaborate to robustly repress cotargets, and suggest important
developmental roles for cotargeting”
(Cherone, Jorgji and Burge 2019, doi:10.1101/gr.249201.119).
It turns out that not only are coding genes often transcribed in an
antisense direction, but there is also a previously unknown “prevalent
antisense transcription” of the genetic sequences giving rise to miRNAs. “We
found that antisense transcripts are tightly regulated, and a substantial
fraction of miRNAs and their antisense transcripts are coexpressed. Sense
miRNAs have been shown to down-regulate the coexpressed antisense
transcripts, whereas the act of antisense transcription, rather than the
transcripts themselves, regulates the expression of sense miRNAs. RNA
editing tends to decrease the miRNA accessibility of the antisense
transcripts, therefore protecting them from being degraded by the
sense-mature miRNAs. Altogether, our study reveals the landscape of
antisense transcription and editing of miRNAs, as well as a previously
unknown reciprocal regulatory circuit of sense–antisense miRNA pairs”
(Song, Li, Yang et al. 2020, doi:10.1101/gr.257121.119).
“In animals, systemic control of metabolism is conducted by metabolic
tissues and relies on the regulated circulation of a plethora of molecules,
such as hormones and lipoprotein complexes. MicroRNAs ... have been widely
associated with the regulation of gene expression in various contexts,
including virtually all aspects of systemic control of metabolism. Here we
focus on glucose and lipid metabolism and review current knowledge of the
role of miRNAs in their systemic regulation. We survey miRNA-mediated
regulation of healthy metabolism as well as the contribution of miRNAs to
metabolic dysfunction in disease, particularly diabetes, obesity and liver
disease. Although most miRNAs act on the tissue they are produced in, it is
now well established that miRNAs can also circulate in bodily fluids,
including their intercellular transport by extracellular vesicles, and we
discuss the role of such extracellular miRNAs in systemic metabolic control
and as potential biomarkers of metabolic status and metabolic disease”
(Agbu and Carthew 2021, doi:10.1038/s41580-021-00354-w).
The following is just a sampling of the extraordinarily wide-ranging repertoire of cellular miRNA activity.
- In “a beautiful example of how microRNAs (miRNAs) can regulate tissue-specific gene expression in a biologically relevant setting”, “Drexel and colleagues ... found that miR-791 is expressed in only three types of carbon dioxide (CO2)-sensing neurons in Caenorhabditis elegans, and its primary function there seems to be repression of two target genes that interfere with the behavioral response to CO2. Interestingly, these two targets are broadly expressed across other tissues. Thus, restricted miRNA expression can lead to target repression in select tissues to promote distinct cellular physiologies” (Pasquinelli 2016, doi:10.1101/gad.290023.116).
- “miRNAs with identical 5' end sequences comprise families and have been proposed to target the same genes. While this may be true in some instances, a role in CO2 sensing was not detected for miR-790, which is identical to miR-791 at positions 2-10 and is expressed in the same neurons. Furthermore, loss of miR-791 alone resulted in misregulation of the akap-1 and cah-3 targets and consequent defective avoidance behavior. Thus, in this biological context, the miR-790/791 family members are not redundant. This in vivo example lends credence to recent biochemical studies that have shown specific target-binding activity by distinct miRNA family members ... Previously, a broad screen for miRNA function indicated that deletion of individual miRNA genes results in no obvious phenotypes, except in rare cases. A current perception is that many miRNAs act redundantly or combinatorially to sculpt gene expression, buffering the loss of individual miRNAs. The study by Drexel et al. (2016) now provides a compelling demonstration that a single miRNA can have a biologically relevant function that may be apparent only in certain contexts. Given the lesson of miR-791, perhaps the idea that miRNAs exert largely redundant fine-tuning functions merits reconsideration” (Pasquinelli 2016, doi:10.1101/gad.290023.116).
- “In Drosophila larvae, mutation of a single microRNA locus affects the animal’s ability to correct its orientation if turned upside down” (blurb in Science for Picao-Osorio, Johnston, Landgraf et al. 2015, doi:10.1126/science.aad0217).
- miRNAs guide protein complexes to specific mRNAs, and elements of those complexes either cleave and destroy the mRNAs or else degrade their translation. Not only can a single miRNA in this way help to regulate hundreds of mRNAs (and, of course, all the proteins that might be produced from those mRNAs), but any given mRNA may have multiple miRNA binding sites recognizable by numerous miRNAs. So miRNAs may achieve a wide array of different, combinatorial effects. Also, a single base difference between two miRNAs can result in their regulating a completely different set of genes and being functionally independent.
- It has been thought that miRNAs bind only to sequences in the 3'-UTR (untranslated region) of mRNAs. But more recent work has shown that miRNAs can directly target the coding regions of mammalian genes. For example, they can inhibit entire families of zinc-finger genes (that is, genes for proteins — very often transcription factors — containing a certain domain referred to as a “zinc finger”) by binding to any one of several coding-sequence repeats within these genes. (Schnall-Levin, Rissland, Johnston et al. 2011; also see Huang, Wu, Ding et al. 2010).
- “We find that sites located in the CDS [coding regions] are most potent in inhibiting translation, while sites located in the 3'-UTR are more efficient at triggering mRNA degradation. Our study suggests that miRNAs may combine targeting of CDS and 3'-UTR to flexibly tune the time scale and magnitude of their post-transcriptional regulatory effects” (Hausser, Syed, Bilen and Zavolan 2013).
- In addition to appropriate 3'-UTR sequences for the miRNA to target, it appears that secondary structure in the 5'-UTR of the target gene is a common prerequisite for mRNA translational repression and subsequent degradation (Meijer, Kong, Lu et al. 2013). “mRNAs that are targeted by miRNA tend to have a higher degree of local secondary structure in their 5' UTR ... [We] found a universal trend of increased mRNA stability near the 5' cap in mRNAs that are regulated by miRNA in animals, but not in plants. Intra-genome comparison showed that gene expression level, GC content of the 5' UTR, number of miRNA target sites, and 5' UTR length may influence mRNA structure near the 5' cap. Our results suggest that the 5' UTR secondary structure performs multiple functions in regulating post-transcriptional processes” (doi:10.1261/rna.042754.113).
- Evidence is also emerging that miRNAs may target the promoter region of genes. It appears that antisense, promoter-associated, noncoding RNA (ncRNA) transcripts lying close to the promoter of the target gene play a role in the process. An miRNA first targets the ncRNA and then a protein associated with the miRNA recruits other protein factors to the promoter, perhaps in order to apply epigenetic “marks” to the chromatin (Younger and Corey 2011).
- By virtue of their pre- and post-transcriptional regulation of mRNAs and their consequent involvement in all aspects of cell biology, miRNAs play an indirect role at multiple levels of gene regulation. For example, by regulating proteins affecting chromatin structure, they influence gene expression at the transcriptional level.
- Pseudogenes (some 19,000 of them, formerly considered functionless products of mutated genes) also have miRNA binding sites, which means they can “compete” with mRNAs and long noncoding RNAs for miRNAs, thereby playing a regulatory role. It appears, in fact, that all RNAs, including those transcribed from pseudogenes, engage in a large-scale, cross-talking regulatory conversation involving their mutual interaction with miRNAs (Salmena, Poliseno, Tay et al. 2011). See “Competing endogenous RNAs” above.
- Depending on conditions, miRNAs can fine-tune many processes, and exert a strong, on-off effect upon others — and this can vary from one cell to the next (Mukherji, Ebert, Zheng et al. 2011). In a substantial proportion of cases, eliminating miRNAs results in no obvious problems — until the organism experiences some sort of stress. This suggests that many miRNAs play a role in buffering against defects and maintaining homeostasis. They sharpen various developmental transitions and, in general, support the organism’s “robustness” — its ability to maintain its functioning in the face of internal or external disturbances. See, for example, Ebert and Sharp 2012; Cassidy, Jha, Posadas et al. 2013.
- “The importance of miRNAs in development has become nearly ubiquitous, with miRNAs contributing to development of most cells and organs. Although miRNAs are clearly interwoven into known regulatory networks that control cell development, the specific modalities by which they intersect are often quite distinct and cleverly achieved. The frequently emerging theme of feed-back and feed-forward loops to either counterbalance or reinforce the gene programs that they influence is a common thread. Many of these examples of miRNAs as developmental regulators are presently found in organs with different miRNAs and targets” (Ivey and Srivastava, doi:10.1101/cshperspect.a008144). An example: “miR-9 and miR-124a modulate STAT3 phosphorylation, which mediates the development of neurons and astrocytes in the brain” (from the journal’s blurb for same article).
- “Interestingly, in contrast to miRNA knockout, miRNA overexpression often leads to specific and easily detectable phenotypes. Indeed, overexpression or misexpression of miRNAs can promote remarkable alterations in cell fate, including dedifferentiation of somatic cells to induced pluripotent stem cells, or transdifferentiation across somatic cell lineages such as fibroblasts to neurons and cardiomyocytes” (Shenoy and Blelloch 2014). This, too, testifies to the normally subtle role of miRNAs, and reminds us that subtle, difficult-to-detect functionality of molecules does not point to their unimportance.
- “MicroRNAs (miRNA) are emerging as critical factors in gene regulation during development; however, their role in adult-onset, age-associated processes is only beginning to be revealed. Here we report that the conserved miRNA miR-34 regulates age-associated events and long-term brain integrity in Drosophila, providing a molecular link between ageing and neurodegeneration. Fly mir-34 expression exhibits adult-onset, brain-enriched and age-modulated characteristics. Whereas mir-34 loss triggers a gene profile of accelerated brain ageing, late-onset brain degeneration and a catastrophic decline in survival, mir-34 upregulation extends median lifespan and mitigates neurodegeneration induced by human pathogenic polyglutamine disease protein” (Liu, Landreh, Cao et al. 2012).
- Mouse research has shown that miRNAs are “essential for the maintenance of the quiescent state” of stem cells (Cheung, Quach, Charville et al. (2012).
- A transcription factor expressed in endothelial cells (inner lining of blood and lymphatic vessels) has a role in protecting against atherosclerosis. It turns out that this transcription factor induces the expression of two miRNAs which in turn are transported to adjacent smooth muscle cells, where they carry out a protective function (Baumann 2012).
- An miRNA role in dampening oscillations in gene expression: “The complexity of multicellular organisms requires precise spatiotemporal regulation of gene expression during development. We find that in the nematode Caenorhabditis elegans approximately 2,000 transcripts undergo expression oscillations synchronized with larval transitions while thousands of genes are expressed in temporal gradients, similar to known timing regulators. By counting transcripts in individual worms, we show that pulsatile expression of the microRNA (miRNA) lin-4 maintains the temporal gradient of its target lin-14 by dampening its expression oscillations. Our results demonstrate that this insulation is optimal when pulsatile expression of the miRNA and its target is synchronous” (Kim, Grün, van Oudenaarden 2013).
- “Morphogens induce biological diversity by operating in a dose-dependent manner. ... microRNAs (miRNAs) are ideally suited to serve the morphogen cause. miRNAs regulate the establishment of morphogen gradients ... by acting on their secretion, distribution and clearance. miRNA are also critical in receiving cells, establishing context-dependency and threshold responses. Moreover, miRNAs contributes to gene networks that transform the graded activity of a morphogen into robust cell fate decisions” (Inui, Montagner and Piccolo 2012).
- miRNAs mature in the cytoplasm and that is where their “canonical” function in targeting mRNAs and regulating translation is carried out. However, the situation now looks more complex, as miRNAs are found to be transported into the nucleus as well. Here they apparently target noncoding RNAs, including long noncoding RNAs and other miRNAs, and modulate their biogenesis and function. “Considering the complexity and diversity of miRNA–target interactions, it is reasonable to imagine that miRNAs and their target ncRNAs form a complex regulatory network within the nucleus. Through this network, miRNAs can control ncRNA homeostasis and balance the tightly regulated, equilibrated state of miRNAs and other ncRNAs” (Chen, Liang, Zhang and Zen 2012).
- There are now “suggestions that miRNA biogenesis may even be occurring locally near synapses and dendrites, an astounding notion that could provide insight into the increased cortical biogenesis that is observed for a number of miRNAs in schizophrenia. ... With a number of other neurological disorders also characterized by miRNA dysfunction, this lends further support to the hypothesis that miRNAs play a fundamental role in regulating the activity-dependent spatiotemporal control of translation at synapses required for long-term potentiation and the homeostatic control of neuronal connectivity. When also considering the large number of primate-specific miRNAs expressed in the brain and that a single pyramidal neuron in the cortex may form up to 10000 synapses with other cells, it is tantalizing to hypothesize a role for miRNA as key regulatory molecules in the development of the exquisitely complex programmes of gene expression and the decentralized modifications of individual synaptodendritic connections that are required for the cortical complexity observed in the human brain” (Carroll, Tooney and Cairns 2013).
- A study was made of human dendritic cells infected with Mycobacterium tuberculosis. About 40% of miRNAs were differentially expressed after infection. “Our findings showed that infection is accompanied by a rapid and strong remodeling of miRNA-mediated regulatory networks, with a shift toward negative miRNA–mRNA correlations. Such a marked shift, largely accounted for by a small number of differentially expressed miRNAs, emphasizes the wide-reaching impact of a subset of miRNAs in the transcriptional response of a cell to infection”. It “seems likely that feedforward and feedback loops are widespread mechanisms in miRNA-mediated regulatory responses in the context of infection” (Siddle, Deschamps, Tailleux et al. 2014).
- Despite expectations of more or less exact, predictable, and digitally precise interactions between so-called “information-bearing molecules”, we’ve been learning in recent years that these molecules relate to each other in a varied, mutually adaptive, and fluid manner. And this is proving true of the relations between miRNA seeds and mRNA target sites. “[Studies] highlight the vast array of non-canonical miRNA-mRNA interactions, and strongly hint that the variability of functional interacting sites is far more extreme than first indicated by comparative genomics studies. It is not unreasonable to consider the possibility that any accessible six nucleotides, whether contiguous in the mRNA or not, is [sic] capable of mediating a functional interaction with a miRNA” (Cloonan 2015a, doi:10.1002/bies.201400191).
- Regarding the miR-183 family of miRNAs (miR-183, miR-96, miR-182): “Normally the expression of the miR-183 cluster is highly specific to the sensory organs and is necessary for sensory development and circadian rhythm ... However, dysregulation of the miR-183 family expression occurs in disorders unrelated to sensory organs. The high expression of these miRs in disease may be permissive or contribute to the altered post-transcriptional landscape in cancer, autoimmune and neurological disorders. Moreover, the individual miR-183 family members cooperate to regulate multiple components of both normal and disease pathways of sensory development, metabolism, apoptosis, DNA repair, metal homeostasis, immune system and circadian rhythm. Coming full circle, these miRs are also regulated by key transcriptional factors that control the above mentioned processes (Dambal, Shah, Mihelich and Nonn 2015, doi:10.1093/nar/gkv703).
- “MicroRNAs imported from the cytoplasm into mitochondria were, surprisingly, found to act as regulators of mitochondrial translation. In turn, translation in mitochondria controls cellular proliferation, and mitochondrial ribosomal subunits contribute to the cytoplasmic stress response. Thus, translation in mitochondria is apparently integrated into cellular processes” (Richter-Dennerlein, Dennerlein, and Rehling 2015, doi:10.1038/nrm4051).
- “Proper functioning of an organism requires cells and tissues to behave in uniform, well-organized ways. How this optimum of phenotypes is achieved during the development of vertebrates is unclear. Here, we carried out a multi-faceted and single-cell resolution screen of zebrafish embryonic blood vessels upon mutagenesis of single and multi-gene microRNA (miRNA) families. We found that embryos lacking particular miRNA-dependent signaling pathways develop a vascular trait similar to wild-type, but with a profound increase in phenotypic heterogeneity. Aberrant trait variance in miRNA mutant embryos uniquely sensitizes their vascular system to environmental perturbations. We discovered a previously unrecognized role for specific vertebrate miRNAs to protect tissue development against phenotypic variability” (Kasper, Moro, Ristori et al. 2017, doi:10.1016/j.devcel.2017.02.021).
-
miRNAs can enhance gene expression
(NOTE: This subsection properly belongs under PRE-TRANSCRIPTIONAL DECISION-MAKING — an illustration of the impossibility of pigeon-holing the significance of elements functioning within an interwoven organic context.)- It has now been shown that some miRNAs are associated with RNA polymerase II and bind to the TATA box in the gene promoters of human peripheral blood mononuclear cells. This is the case, for example, with the interleukin-2 (IL-2) gene in CD4+ T-lymphocytes, with the result that the IL-2 mRNA and protein production are elevated. “Through direct interaction with the TATA-box motif, [the miRNA sequence] let-7i facilitates the PIC assembly and transcription initiation of IL-2 promoter. Several other cellular miRNAs, such as mir-138, mir-92a or mir-181d, also enhance the promoter activities via binding to the TATA-box motifs of insulin, calcitonin or c-myc, respectively ... our data demonstrate that the interaction with core transcription machinery is a novel mechanism for miRNAs to regulate gene expression” (Zhang, Fan, Zhang et al. 2014, doi:10.1261/rna.045633.114).
- “Since the binding between miRNA and TATA-box motif is sequence specific, we believe that the regulation of transcription by miRNAs is much more specific and accurate than that by protein transcription factors. Accumulating evidence has demonstrated that the biogenesis and function of miRNAs are also regulated by many transduction signals (Cullen 2004; O'Donnell et al. 2005; Krol et al. 2010). Therefore, our findings indicate a novel signal pathway to specifically regulate the gene expression at transcriptional level. This is in accordance with the observation that a number of miRNAs are found in the nucleus” (Zhang, Fan, Zhang et al. 2014, doi:10.1261/rna.045633.114).
- A study connecting long noncoding RNAs, microRNAs, chromatin remodeling, and gene activation: “We explore the function of lncRNAs in small RNA-triggered transcriptional gene activation (TGA), a process in which microRNAs (miRNAs) or small interfering RNAs (siRNAs) associated with Argonaute (Ago) proteins induce chromatin remodeling and gene activation at promoters with sequence complementarity ... we demonstrated that small RNA-triggered TGA occurs at sites where antisense lncRNAs are transcribed through the reporter gene and promoter. Small RNA-induced TGA coincided with the enrichment of Ago2 at the promoter region, but Ago2-mediated cleavage of antisense lncRNAs was not observed ... Termination of nascent antisense lncRNAs abrogated gene activation triggered by small RNAs, and only allele-specific cis-acting antisense lncRNAs, but not trans-acting lncRNAs, were capable of rescuing TGA. Hence, this model revealed that antisense lncRNAs can mediate TGA in cis and not in trans, serving as a molecular scaffold for a small RNA–Ago2 complex and chromatin remodeling” (Zhang, Li, Burnett and Rossi 2014; doi:10.1261/rna.043968.113).
-
Role of Argonaute proteins
- microRNAs become part of ribonucleoprotein assemblies known as miRISCs (miRNA-induced silencing complexes), a primary component of which is one or another member of the Argonaute (“AGO”) protein family. This protein plays a major role in degrading or repressing the translation of the mRNAs to which it is guided by the associated miRNA. There is huge context-specific regulatory potential in the variable constitution of the miRISC. “The capacity for a large number of other proteins to associate with the AGO core of miRISCs introduces the potential for many different miRISCs to exist within a cell at any given time”. There are four distinct AGO proteins “with specific expression patterns, subcellular localizations, protein-binding partners, and biochemical capabilities”. For example: researchers “have identified a significant decrease in AGO1 — but not AGO2 — expression in a number of tumour cell lines, and also observed an AGO1-specific increase in expression levels throughout neuronal differentiation” (Carroll, Tooney and Cairns 2013).
- It has been thought that miRNAs are wholly responsible for guiding Argonaute proteins to the target mRNAs. However, new research seems to verify the hypothesis that “AGO has its own binding preference within target mRNAs, independent of guide miRNAs. ... We have identified a structurally accessible and evolutionarily conserved region (~10 nucleotides in length) that alone can accurately predict AGO-mRNA associations, independent of the presence of miRNA binding sites. ... These findings reveal a novel function of AGOs as sequence-specific RNA-binding proteins, which may aid miRNAs in recognizing their targets with high specificity” (Li, Kim, Nutiu et al. 2014).
- “miRNAs are enclosed within Argonaute (Ago) proteins”, and post-translational control of these proteins “can relay upstream stimuli to downstream gene regulatory responses, in contexts that range from hypoxia and cell differentiation to antiviral defense”. In particular, “A growing theme of recent years is how post-translational modifications of Ago proteins, such as prolyl hydroxylation, phosphorylation, ubiquitination, and poly-ADP-ribosylation, alter miRNA activity at global or specific levels”. Thus, the varied modifications of Ago proteins take their place within a larger picture of remarkable fluidity and contextual sensitivity. This picture includes “(i) diverse post-transcriptional modifications of small RNA intermediates, mature miRNAs, or the mRNAs encoding miRNA pathway factors; (ii) post-translational modifications of miRNA pathway factors; and (iii) the action of ancillary proteins that modulate the core miRNA machinery” (Jee and Lai 2014).
- “A conserved phosphorylation site in Argonaute 2, a key effector of miRNA‐dependent gene regulation, controls mRNA binding in human and worms, revealing that the activity of the RNAi machinery is dynamically regulated” (Huberdeau, Zeitler, Hauptmann 2017, doi:10.15252/embj.201696386).
-
Role of other proteins
[This applies to siRNAs also. See
Small interfering RNAs below.]
Many other proteins play a role in the biogenesis and regulation of miRNAs, as indicated in the next section. These proteins themselves are subject to complex regulation. For example, taking just one type of molecule, the human Ago proteins: “The activity of Agos can be modulated through post-translational modifications including proline hydroxylation, which increases slicing activity; SUMOylation, which increases protein stability; ADP ribosylation, which relieves both slicing and translation repression; and phosphorylation, which can either enhance or inhibit silencing efficacy” (Ipsaro and Joshua-Tor 2015, doi:10.1038/nsmb.2931). -
microRNAs are themselves subject to extensive regulation
“miRNA biogenesis is regulated at multiple levels, including at the
level of miRNA transcription; its processing by Drosha and Dicer in the
nucleus and cytoplasm, respectively; its modification by RNA editing,
RNA methylation, uridylation and adenylation; Argonaute loading; and
RNA decay. Non-canonical pathways for miRNA biogenesis, including those
that are independent of Drosha or Dicer, are also emerging” (Ha and Kim
2014).
“Regulation of miRNA expression can occur both at the
transcriptional level and at the post-transcriptional level during
miRNA processing. Recent studies have elucidated specific aspects
of the well-regulated nature of miRNA processing involving various
regulatory proteins, editing of miRNA transcripts, and cellular
localization. In addition, single nucleotide polymorphisms in miRNA
genes can also affect the processing efficiency of primary miRNA
transcripts” (Slezak-Prochazka, Durmus, Kroesen and van den Berg
2010).
“Upstream of miRNAs is a network of cell type-specific transcription
factors that function together with epigenetic regulators to tightly
regulate miRNA levels spatially and temporally. miRNA levels are
further fine-tuned through post-transcriptional mechanisms that
regulate their processing to the mature form, as well as their
stability. Downstream of miRNAs are large networks of mRNA targets
that influence cell fate choice. The ultimate effect of miRNAs on those
targets is influenced by several factors. First, the number of miRNA
binding sites within each target, which can be regulated by APA
[alternative polyadenylation]. Second, whether other miRNAs target the
same transcript. Moreover, RBPs [RNA-binding proteins] may bind along
with miRNAs either synergizing or antagonizing the activity of the
associated miRNAs. Third, multiple RNAs may be competing for the same
miRNA, raising the possibility that a small number of these transcripts
titrate away the miRNAs from other potential targets”
(Shenoy and Blelloch 2014).
“Argonaute (Ago) proteins interact with various binding partners and play
a pivotal role in microRNA (miRNA)-mediated silencing pathways ... we
identified a putative RNA-binding protein FAM120A (also known as
OSSA/C9ORF10) as an Ago2 interacting protein ... FAM120A binds to
homopolymeric tracts in 3′-UTRs of about 2000 mRNAs, particularly poly(G)
sequences ... greater than one-third of mRNAs bound by Ago2 in mESCs are
co-bound by FAM120A. Furthermore, such FAM120A-bound Ago2 target genes
are not subject to Ago2-mediated target degradation. Reporter assays
suggest that the 3′-UTRs of several FAM120A-bound miRNA target genes are
less sensitive to Ago2-mediated target repression than those of
FAM120A-unbound miRNA targets and FAM120A modulates them via its G-rich
target sites. These findings suggest that Ago2 may exist in multiple
protein complexes with varying degrees of functionality”
(Kelly, Suzuki, Zamudio et al. 2019, doi:10.1261/rna.071621.119)
“Binding of microRNAs (miRNAs) to mRNAs normally results in
post-transcriptional repression of gene expression. However, extensive
base-pairing between miRNAs and target RNAs can trigger miRNA
degradation, a phenomenon called target RNA-directed miRNA degradation
(TDMD) ... [We] identified numerous candidate TDMD triggers, focusing on
their ability to induce nontemplated nucleotide addition at the miRNA 3′
end. When exogenously expressed in various cell lines, eight triggers
induce degradation of corresponding miRNAs. Both the TDMD base-pairing
and surrounding sequences are essential for TDMD ... Furthermore,
degradation of miR-221 and miR-222 by a trigger in BCL2L11, which
encodes a proapoptotic protein, enhances apoptosis. Therefore, we
uncovered widespread TDMD triggers in target RNAs and demonstrated an
example that could functionally cooperate with the encoded protein
(Li, Sheng, Li et al. 2021, doi:10.1101/gad.348874.121).
- miRNA variants of different sorts (isomiRs) are now known to exist, and are thought to have “broad implications in mRNA targeting, stability and/or gene expression regulation” (Pantano, Lorena, Estivill, and Martí 2010.) For example, fruit fly research suggests that “subtle variability in isomiR expression ... is regulated and biologically meaningful,” and plays a role in gene regulation especially during embryonic development, but also in adult tissues (Fernandez-Valverde, Taft and Mattick 2010). And, from a more recent article: there is “a growing appreciation for the fact that individual miRNAs can be heterogeneous in length and/or sequence. These variants...can be expressed in a cell-specific manner, and numerous recent studies suggest that at least some isomiRs may affect target selection, miRNA stability, or loading into the RNA-induced silencing complex (RISC)” (Neilsen, Goodall and Bracken 2012).
- One way isomiRs can be regulated (apart, say, from RNA editing), is by having their 5' or 3' ends shifted by a few nucleotides relative to the corresponding unmodified miRNAs. Xia and Zhang (2014) performed an extensive study of 5-'isomiRs in human, mouse, fruitfly, and worm. “The analysis has revealed robustness and plasticity of miRNA mediated post-transcriptional gene regulation. Though they shared a substantial amount of common target genes, many 5'-isomiRs and [their associated, unmodified] miRNAs also had their distinct exclusive target genes”. The overall results of the study “revealed a broad existence of 5'-isomiRs in the four species, many of which were conserved and could arise from genomic loci of canonical and non-canonical miRNAs”. The 5'-isomiRs varied across tissues and were associated with distinctive structural elements in the RNAs from which they were derived. “Eighteen 5'-isomiRs had aberrant expression in psoriatic human skin, suggesting their potential function in psoriasis pathogenesis”.
- Regarding breast cancer: “We report that the full isomiR profiles, from both known and novel human-specific miRNA loci, are particularly rich in information and can distinguish tumor from normal tissue much better than the archetype miRNAs. IsomiR expression is also dependent on the patient's race, exemplified by miR-183-5p, several isomiRs of which are upregulated in triple negative breast cancer in white but not black women. Additionally, we find that an isomiR's 5' endpoint and length, but not the genomic origin, are key determinants of the regulation of its expression ... Each isomiR has a distinct impact on the cellular transcriptome” (Telonis, Loher, Jing et al. 2015, doi:10.1093/nar/gkv922).
-
Messenger RNAs (mRNAs) that are down-regulated by miRNAs typically
associate with many RNA-binding proteins carrying out numerous
regulatory functions. Some of these proteins increase and others
decrease the effectiveness of miRNA action against the given mRNA.
So these proteins represent an additional level of regulation of
miRNA activity, and “binding sites of miRNAs and RNA-BPs [RNA
binding proteins] should be considered in combination when
interpreting and predicting miRNA regulation in vivo” (Jacobsen,
Wen, Marks and Krogh 2010).
- An example: an RNA-binding protein can bind to the 3' untranslated region of an mRNA and thereby alter its local shape, with the result that miRNAs gain greater access to the mRNA and downregulate its translation (Kedde, Kouwenhove, Zwart et al. 2010).
- The Dicer protein cleaves pre-miRNA molecules in the cytoplasm to form miRNAs, and therefore is a key player in the production of numerous miRNAs. Both Dicer and the pre-mRNA must be exported from the nucleus in order for Dicer to do its work, and this export depends on the exportin-5 protein, which is a limiting factor. The nucleus of human cells also requires exportin-5 protein. Therefore “overexpression of a substrate miRNA is able to saturate the exportin-5 export pathway. This leads to a decreased association between exportin-5 and its other substrates — such as the Dicer mRNA, which results in reduced amounts of Dicer protein in the cell. Thus, there is cross-regulation between pre-miRNAs and their processing enzyme, Dicer, which could help balance the amounts of the enzyme and its substrate” (Riddihough 2011; Bennasser et al. 2011).
- An alternatively spliced protein known as loquacious (loqs) partners with Dicer in three distinct isoforms, with different effects. In fruit flies two of the protein forms, in conjunction with Dicer, generate known types of siRNA. “Surprisingly [a third form of the protein] tunes where Dicer-1 cleaves pre-miR-307a, generating a longer miRNA isoform with a distinct seed sequence and target specifity”. A mammalian homolog to loqs “similarly tunes where Dicer cleaves pre-miR-132. Thus, Dicer-binding partner proteins change the choice of cleavage site by Dicer, producing miRNAs with target specificities different from those made by Dicer alone or Dicer bound to alternative protein partners” (Fukunaga, Han, Hung et al. 2012).
- While Dicer plays a major regulatory role in generating miRNAs, it turns out that an miRNA in turn can play a major role in regulating Dicer. In particular, “during zebrafish hindbrain development dicer expression levels are controlled by miR-107 to tune the biogenesis of specific miRNAs, such as miR-9, whose levels regulate neurogenesis” (Ristori, Lopez-Ramirez, Narayanan et al. 2015, doi:10.1016/j.devcel.2014.12.013).
-
The depth of intertwined regulatory processes is indicated in the
accompanying figure from an article entitled “The Many Faces of Dicer:
The Complexity of the Mechanisms Regulating Dicer Gene Expression and
Enzyme Activities” (Kurzynska-Kokorniak, Koralewska, Pokornowska et
al. 2015, doi:10.1093/nar/gkv328 ). The Dicer protein is just one
element in the regulation of miRNAs, yet it is enmeshed in a dense
network of other elements bearing on its own functioning.
![[Image: some factors
in the regulation of Dicer]](./Images/dicer_regulation.jpg "Some factors in the regulation of
Dicer")
- “Autophagy [a process by which a cell degrades its own cellular components] regulates microRNA (miRNA) biogenesis by fine-tuning the levels of the miRNA machinery components DICER and argnonaute (AGO). ... Autophagy does not serve to degrade miRNAs (bound to DICER and AGO) but instead to stabilize their abundance” (David 2013).
- Nucleotides that are not templated by the DNA from which the miRNAs arose are now known to be added as “tails” to precursor and mature miRNAs. The most common addition is uridine, but adenosine and cytidine may also be added. The functional implications of these additions are only beginning to be investigated, but it appears in some cases that the presence of a tail contributes to the degradation of the miRNA (Newman, Mani and Hammond 2011).
- Actually, so far as uridine is concerned, whereas a long, single-stranded tail in at least some cases inhibits the processing of premature miRNAs into mature, functional ones, mono-uridylation (addition of just a single uridine) promotes miRNA maturation (David 2012). And so “nontemplated nucleotide addition represents a further layer of complexity for the regulation of miRNA production and activity” (Newman, Mani and Hammond 2011).
- Dimethylation (addition of two methyl groups) has been found to occur at the head (5' end) of premature miRNAs, with a negative effect on miRNA biogenesis (David 2012).
- More generally, the termini (both “head” and “tail”) of miRNAs are modified in ways only beginning to be understood. “Analysis in Drosophila revealed multiple modification patterns, including select alterations of 5' termini, many 3' resection events, and unexpectedly abundant 3' untemplated monouridylation... Strikingly, we found many mirtrons [intron-derived miRNAs] whose modified reads are more abundant than those produced by primary processing...Altogether, these findings substantially broaden the complexity of terminal modification pathways acting upon small regulatory RNAs” (Westholm, Ladewig, Okamura et al. 2012).
- miRNAs sometimes occur in clusters in introns. These clusters were thought to be expressed in conjunction with transcription of the parent gene. However, researchers are now verifying instances in humans where intronic miRNAs are separately transcribed — that is, independently of the parent gene. And whereas it was previously thought that the miRNAs of a cluster were always expressed together, it now appears that sometimes the individual miRNAs of a cluster are independently expressed, with alternative splicing playing a role in the selection process (Ramalingam, Palanichamy, Singh et al. 2014).
- “Sequence heterogeneity at the ends of mature microRNAs (miRNAs) is well documented, but its effects on miRNA function are largely unexplored. Here we studied the impact of miRNA 5'-heterogeneity, which affects the seed region critical for target recognition. Using the example of miR-142-3p, an emerging regulator of the hematopoietic lineage in vertebrates, we show that naturally coexpressed 5'-variants (5'-isomiRs) can recognize largely distinct sets of binding sites. Despite this, both miR-142-3p isomiRs regulate exclusive and shared targets involved in actin dynamics. Thus, 5'-heterogeneity can substantially broaden and enhance regulation of one pathway. Other 5'-isomiRs, in contrast, recognize largely overlapping sets of binding sites. This is exemplified by two herpesviral 5'-isomiRs that selectively mimic one of the miR-142-3p 5'-isomiRs. We hypothesize that other cellular and viral 5'-isomiRs can similarly be grouped into those with divergent or convergent target repertoires, based on 5'-sequence features” (Manzano, Forte, Raja et al. 2015, doi:10.1261/rna.048876.114).
- “While many neuronal miRNAs were previously shown to modulate neuronal morphogenesis, little is known regarding the regulation of miRNA function ... we identified two novel regulators of neuronal miRNA function, Nova1 and Ncoa3. Both proteins are expressed in the nucleus and the cytoplasm of developing hippocampal neurons. We found that Nova1 and Ncoa3 stimulate miRNA function by different mechanisms that converge on Argonaute (Ago) proteins, core components of the miRNA‐induced silencing complex (miRISC). While Nova1 physically interacts with Ago proteins, Ncoa3 selectively promotes the expression of Ago2 at the transcriptional level. We further show that Ncoa3 regulates dendritic complexity and dendritic spine maturation of hippocampal neurons in a miRNA‐dependent fashion” (Störchel, Thümmler, Siegel 2015, doi:10.15252/embj.201490643).
- MicroRNA-122 (miR-122) is expressed at high levels in hepatocytes. It is selectively stabilized via polyadenylation by the cytoplasmic poly(A) polymerase GLD-2, and destabilized via deadenylation by poly(A)-specific ribonuclease (PARN). In addition “CUG-binding protein 1 (CUGBP1) specifically interacts with miR-122 and other UG-rich miRNAs, and promotes their destabilization”. CUGBP1 is thought to recruit PARN to miR-122. “These results indicate that the cellular level of miR-122 is determined by the balance between the opposing effects of GLD-2 and PARN/CUGBP1 on the metabolism of its 3'-terminus” (Katoh, Hojo and Suzuki 2015, doi:10.1093/nar/gkv669).
- “Under specific conditions, abundant and highly complementary target RNAs can trigger miRNA degradation by a mechanism involving nucleotide addition and exonucleolytic degradation ... We report here that both the degree of complementarity and the ratio of miRNA/target abundance are crucial for the efficient decay of the small RNA ... we set [out] to identify the [protein] factors involved in target-mediated miRNA degradation. Among the retrieved proteins, we identified members of the RNA-induced silencing complex, but also RNA modifying and degradation enzymes. We show that [the Perlman Syndrome 3'-5' exonuclease DIS3L2] interacts with Argonaute 2 and functionally validate its role in target-directed miRNA degradation” (Haas, Cetin, Mélanie Messmer et al. 2016, doi:10.1093/nar/gkw040).
- “Alterations in the balance of mRNA and microRNA (miRNA) expression profiles contribute to the onset and development of colorectal cancer. The regulatory functions of individual miRNA-gene pairs are widely acknowledged, but group effects are largely unexplored. We performed an integrative analysis of mRNA–miRNA and miRNA–miRNA interactions ... This investigation resulted in a hypernetwork-based model, whose functional backbone was fulfilled by tight micro-societies of miRNAs. These proved to modulate several genes that are known to control a set of significantly enriched cancer-enhancer and cancer-protection biological processes, and that an array of upstream regulatory analyses demonstrated to be dependent on miR-145, a cell cycle and MAPK signaling cascade master regulator. In conclusion, we reveal miRNA-gene clusters and gene families with close functional relationships and highlight the role of miR-145 as potent upstream regulator of a complex RNA–RNA crosstalk, which mechanistically modulates several signaling pathways and regulatory circuits that when deranged are relevant to the changes occurring in colorectal carcinogenesis” (Mazza, Mazzocolli, Fusilli et al. 2016, doi:10.1093/nar/gkw245).
- “We show that a genome-encoded transcript harboring a near-perfect and deeply conserved miRNA-binding site for miR-29 controls zebrafish and mouse behavior ... The miR-29-binding site is located within the 3′ UTR. We show that the near-perfect miRNA site selectively triggers miR-29b destabilization through 3′ trimming and restricts its spatial expression in the cerebellum. Genetic disruption of the miR-29 site within mouse Nrep results in ectopic expression of cerebellar miR-29b and impaired coordination and motor learning. Thus, we demonstrate an endogenous target-RNA-directed miRNA degradation event and its requirement for animal behavior” (Bitetti, Mallory, Golini et al. 2018; doi:10.1038/s41594-018-0032-x).
- “We show here in C. elegans that nitric oxide derived from resident bacteria promotes widespread S-nitrosylation of the host proteome. We further show that microbiota-dependent S-nitrosylation of C. elegans Argonaute protein (ALG-1) — at a site conserved and S-nitrosylated in mammalian Argonaute 2 (AGO2) — alters its function in controlling gene expression via microRNAs. By selectively eliminating nitric oxide generation by the microbiota or S-nitrosylation in ALG-1, we reveal unforeseen effects on host development. Thus, the microbiota can shape the post-translational landscape of the host proteome to regulate microRNA activity, gene expression, and host development” (Seth, Hsieh, Jamal et al. 2019, doi:10.1016/j.cell.2019.01.037).
-
Role of intercellular and exogenous microRNAs
“Exosomes and other small extracellular vesicles (sEVs) [not to be
confused with RNA degradation complexes also known as “exosomes”] provide
a unique mode of cell-to-cell communication in which microRNAs (miRNAs)
produced and released from one cell are taken up by cells at a distance
where they can enact changes in gene expression. However, the mechanism
by which miRNAs are sorted into exosomes/sEVs or retained in cells
remains largely unknown. Here we demonstrate that miRNAs possess sorting
sequences that determine their secretion in sEVs (EXOmotifs) or cellular
retention (CELLmotifs) and that different cell types, including white and
brown adipocytes, endothelium, liver and muscle, make preferential use of
specific sorting sequences, thus defining the sEV miRNA profile of that
cell type. Insertion or deletion of these CELLmotifs or EXOmotifs in a
miRNA increases or decreases retention in the cell of production or
secretion into exosomes/sEVs. Two RNA-binding proteins, Alyref and Fus,
are involved in the export of miRNAs carrying one of the strongest
EXOmotifs, CGGGAG. Increased miRNA delivery mediated by EXOmotifs leads
to enhanced inhibition of target genes in distant cell”
(Garcia-Martin, Wang, Brandão et al. 2022,
doi:10.1038/s41586-021-04234-3).
- “Overwhelming evidence is also now accumulating to support hypotheses for miRNA and other small non-coding RNA molecules in autocrine, paracrine, and exocrine signalling events” that operate between cells. miRNAs “can even be taken up into recipient cells to mediate silencing effects” (Carroll, Tooney and Cairns 2013).
- The plant miRNA known as miR-168, abundant in rice, has “recently been shown to be present in human blood plasma. This investigation also revealed plant miRNA to be stable in cooked foods, with dietary consumption of plant material resulting in exogenous plant miRNAs being absorbed into the bloodstream of mice from the gastrointestinal tract. Plant miR-168 was even shown to regulate the expression levels of target genes in the liver such as LDLRAP1 (low-density lipoprotein receptor adapter protein 1), resulting in decreased LDL removal from blood plasma in mice. Indeed, such a significant discovery revolutionizes the complexity with which miRNAs are considered to function in mammals throughout various developmental and pathophysiological processes” (Carroll, Tooney and Cairns 2013).
- “microRNAs (miRNAs) are important regulators of gene expression. Although they mostly act in the cells that produce them, they can also be exchanged between cells and even between organisms. In plants, miRNAs can be exchanged between the host plant and certain parasites, thereby modulating the parasitic relationship, but whether plants use such ‘exogenous’ miRNAs more broadly has been elusive. Betti et al. now show that plants can take up miRNAs generated by other plants, and that these exogenous miRNAs are active in silencing the expression of their target genes” (Strzyz 2021, doi:10.1038/s41580-021-00431-0).
-
Role of microRNA precursors
- It’s been found that miRNA precursors — pri-mRNA and pre-mRNA — can compete with the corresponding mature miRNA for binding to targets. The authors of one study conclude: “Altogether, it is likely that precursor miRNAs competing with mature miRNAs and affecting their activity constitute a common regulatory event that could more precisely define important regulatory roles in development and cancer. ... This model offers additional layers of control over existing mechanisms such as miRNA sponges or modifications in miRNA seed regions because the effect of miRNA precursors in gene regulation is target specific. Moreover, because the mature forms are generated from the intermediate precursors, this mechanism can be tightly regulated and can couple or decouple expression from activity of the miRNA to result in differential regulation of mRNAs containing the same MRE [miRNA-response element] target. When taken together, these types of scenarios may further explain why miRNA levels and target-gene repression are not always correlated ... Our results challenge the dogma in which miRNA precursors are considered to be mere nonfunctional intermediates in the miRNA biogenesis pathway” (Roy-Chaudhuri, Valdmanis, Zhang et al. 2014).
-
Small interfering RNAs (siRNAs)
Small interfering RNAs, like miRNAs (see preceding section, much of which
applies in one way or another to siRNAs as well), are small RNA molecules
that target mRNAs, preventing their translation into protein and thereby
playing a vast role in regulation of gene expression. However, siRNAs are
more precisely targeted (that is, have fewer targets, more exactly
identified) than miRNAs. As with miRNAs, the role of siRNAs is being found
to extend beyond the targeting of cytoplasmic mRNAs for degradation, and
into the nucleus.
Small interfering RNAs (and the closely related piwi-interacting RNAs
discussed immediately below) show a remarkable variety of performance.
For example, regarding their transposon-silencing activity: “RNA silencing
pathways recognize several distinguishing features of transposons,
including their tendency to produce dsRNA, exist in unusual chromosomal
arrangements, exhibit suboptimal gene expression properties, and occupy
specialized chromatin contexts. The extent to which these features are
sufficient to distinguish transposons from host genes is unknown. In this
regard, it is interesting to note that distinct RNA silencing pathways may
act combinatorially to identify non-self elements. ... Recent observations
further suggest that the specificity of RNA silencing pathways for
transposons may involve not only distinguishing signals in the transposons
themselves, but also protective signals possessed by host genes” (Dumesic
and Madhani 2014).
“RNA interference (RNAi) is a major, powerful platform for gene
perturbations, but is restricted by off-target mechanisms. Communication
between RNAs, small RNAs, and RNA-binding proteins (RBPs) is a pervasive
feature of cellular RNA networks. We present a crosstalk scenario,
designated as crosstalk with endogenous RBPs’ (ceRBP), in which small
interfering RNAs or microRNAs with seed sequences that overlap RBP motifs
have extended biological effects by perturbing endogenous RBP activity.
Systematic analysis of small interfering RNA (siRNA) off-target data and
genome-wide RNAi cancer lethality screens using 501 human cancer cell lines,
a cancer dependency map, identified that seed-to-RBP crosstalk is
widespread, contributes to off-target activity, and affects RNAi
performance”
(Suzuki, Spengler, Grigelioniene et al. 2018, doi:10.1038/s41588-018-0104-1).
- siRNAs play an important role in gene regulation by structuring chromatin — particularly aiding in the formation of heterochromatin.
- “Epigenetic modifications directed by small RNAs [including siRNAs] have been shown to cause transcriptional repression in plants, fungi and animals.” “Across organisms, nuclear RNAi [RNA interference, here involving both siRNA and piRNA] predominantly operates at heterochromatic loci, where it facilitates sequence-specific silencing through the direction of histone H3K9 methylation and/or cytosine methylation” (Castel and Martienssen 2013).
- Outside constitutive heterochromatin, “increasing evidence indicates that RNAi [including siRNA activity] regulates transcription through interaction with transcriptional machinery” (Castel and Martienssen 2013). Repression of gene expression then occurs by preventing transcription rather than via post-transcriptional regulation. For example: “Most genes and gene promoters appear to be transcribed to some extent and experimental observations suggest that non-coding RNAs interact with target loci via Watson–Crick-based RNA:RNA hybridization and not by double-stranded DNA invasion. Temporal studies have determined that exogenously introduced siRNAs targeted to a promoter region interact first with Argonautes 1 and 2 (AGO1 and AGO2). siRNA and AGO interactions is found within the first 24 h, at the siRNA targeted promoter and is followed shortly thereafter with the recruitment of the H3K9me2 and H3K27me3 silent state epigenetic marks, and later by the recruitment of DNA methyltransferase and DNA methylation at 72-96 h for some genes ... a key consistent feature [of the relevant studies] has been the observations that promoter-directed small RNAs can modulate gene transcription and that some level of epigenetic based silencing is ongoing in the observed silenced genes” (Weinberg and Morris 2016, doi:10.1093/nar/gkw139).
- Regarding how small RNA-loaded Argonaute protein complexes target chromatin to mediate silencing: “Using fission yeast, we demonstrate that transcription of the target locus is essential for RNA-directed formation of heterochromatin. However, high transcriptional activity is inhibitory; thus, a transcriptional window exists that is optimal for silencing. We further found that pre-mRNA splicing is compatible with RNA-directed heterochromatin formation. However, the kinetics of pre-mRNA processing is critical. Introns close to the 5' end of a transcript that are rapidly spliced result in a bistable response whereby the target either remains euchromatic or becomes fully silenced. Together, our results discount siRNA–DNA base pairing in RNA-mediated heterochromatin formation” (Shimada, Mohn and Bühler 2016, doi:10.1101/gad.292599.116).
-
Piwi-interacting RNAs (piRNAs)
These form another class of small RNAs (26 – 31 nucleotides). Our
understanding of them is still fragmentary, but rapidly developing. They
associate with piwi proteins, analogously to the association of
miRNAs with Argonaut proteins in the RISC complex. The piwi protein is in
fact a member of the Argonaut family. (This section should be much
larger.)
“The PIWI-interacting RNA (piRNA) pathway protects genome integrity in part
through establishing repressive heterochromatin at transposon loci.
Silencing requires piRNA-guided targeting of nuclear PIWI proteins to
nascent transposon transcripts, yet the subsequent molecular events are not
understood. Here, we identify SFiNX (silencing factor interacting nuclear
export variant), an interdependent protein complex required for
Piwi-mediated cotranscriptional silencing in Drosophila. SFiNX
consists of Nxf2–Nxt1, a gonad-specific variant of the heterodimeric
messenger RNA export receptor Nxf1–Nxt1 and the Piwi-associated protein
Panoramix. SFiNX mutant flies are sterile and exhibit transposon
derepression because piRNA-loaded Piwi is unable to establish
heterochromatin. Within SFiNX, Panoramix recruits heterochromatin effectors,
while the RNA binding protein Nxf2 licenses cotranscriptional silencing”
(Batki, Schnabl, Wang et al. 2019, doi:10.1038/s41594-019-0270-6).
“Since their discovery as a germline-specific defense mechanism against TEs
[transposable elements], piRNAs and PIWI proteins have been involved in a
wide range of regulatory processes through the regulation of non-TE mRNAs.
Because they were shaking up ‘dogma’, these PIWI/piRNA functions unrelated
to TEs have been reluctantly accepted. However, it is unsurprising that, as
an efficient system for mRNA decay, the piRNA pathway has been repurposed
for cellular mRNA regulation. It is striking that the same developmental
programs are regulated by piRNAs and PIWI proteins in very distant species.
Biological functions of piRNA/PIWI-dependent mRNA regulation include
germline development and specific processes linked to sexual reproduction
such as sex determination and dosage compensation. Massive maternal mRNA
decay during the MZT [maternal-to-zygotic transition] in early embryos also
depends on piRNAs and PIWI proteins in both Drosophila and
Aedes ... A challenge for future studies is to understand how mRNA
regulation by piRNAs and PIWI proteins is conserved although not at the
sequence level. In contrast to miRNAs whose sequences are conserved through
evolution, those of the piRNAs are not — even between closely related
species. piRNAs are extremely diverse and their pool can evolve rapidly
within a species. In addition, the low level of base pairing required to
target mRNAs indicates that most mRNAs might potentially be regulated by
piRNAs. How is selectivity achieved? Cooperation with other regulatory
pathways (e.g., RNA-binding proteins, mRNA modifications) are likely to
contribute strongly. Consistent with this, piRNA-based regulation is often
redundant with other regulatory mechanisms and involved in the fine-tuning
of developmental processes ... Positive mechanisms of action of piRNAs and
PIWI proteins in mRNA stabilization and translational activation have been
discovered ... In addition, PIWI proteins act independently of piRNAs in
cancer progression and metastasis ... Future analyses are likely to identify
additional biological functions of piRNAs and PIWI proteins, particularly
ones linked to germline development and sexual reproduction”
(Ramat and Simonelig 2021, doi:10.1016/j.tig.2020.08.011).
- Piwi-interacting RNAs vary a great deal in sequence and function among species, and their role has been difficult to tie down. In both the germline and gonadal somatic cells of mammals they appear to play an important role, especially during embryogenesis, in silencing of transposon “genes” by cleaving the transcripts expressed from these genes.
- “The piRNA pathway has other essential functions in germline stem cell maintenance and in maintaining germline DNA integrity,” and has also been found to play a role in maternal RNA decay affecting embryonic development of the head in Drosophila (Rouget, Papin, Boureux et al. 2010).
- “There is emerging evidence that some piRNAs may also target protein-coding genes in both the germline and the soma. In addition, piRNAs affect chromatin structure and transcription through effects on de novo methylation at loci containing transposable elements” (separate authors’ summary for Siomi, Sato, Pezic and Aravin 2011).
- In this connection there is a bit of a paradox: piRNAs affect chromatin structure by applying “repressive” epigenetic marks, thereby inhibiting expression of transposable elements. But piRNAs are derived from the very DNA sequences that must be repressed, and therefore those sequences must be actively expressed in order to give rise to the repressive function of the piRNAs (Olovnikov, Aravin and Toth 2012).
-
Small intronic transposable element RNAs (siteRNAs)
- In a study on frogs: “We identify a new class of small noncoding RNAs that we name siteRNAs, which align in clusters to introns of protein-coding genes. We show that siteRNAs are derived from remnants of transposable elements present in the introns. We find that genes containing clusters of siteRNAs are transcriptionally repressed as compared with all genes. Furthermore, we show that this is true for individual genes containing siteRNA clusters, and that these genes are enriched in specific repressive histone modifications. Our data thus suggest a new mechanism of siteRNA-mediated gene silencing in vertebrates, and provide an example of how mobile elements can affect gene regulation” (Harding, Horswell, Heliot et al. 2014).
- “Our work shows that siteRNA clusters coincide with the deposition of repressive epigenetic marks to effect transcriptional repression in the early vertebrate embryo of groups of genes characterized by specific transposable element remnants in their introns. We suggest that the siteRNAs act predominantly in a cis [local] mechanism, being both produced as a result of transcription of the transposable element remnants, and acting as guides to modify chromatin structure” (Harding, Horswell, Heliot et al. 2014).
-
Stable Intronic Sequence RNAs (sisRNAs)
“Intronic sequences are often regarded as ‘nonsense’ transcripts that are
rapidly degraded. We highlight here recent studies on intronic sequences
that play regulatory roles as long noncoding RNAs (lncRNAs) which are
classified as sisRNAs. Interestingly, sisRNAs come in different forms and
are produced via a variety of ways. They regulate genes at the DNA, RNA,
and protein levels, and frequently engage in autoregulatory feedback loops
to ensure cellular homeostasis under normal and stress conditions.”
“sisRNAs can be produced through splicing-dependent/independent pathways,
and interact with satellite bodies for their decay.”
“[sisRNAs] can either repress or enhance gene expression through feedback
loops or affect splicing events by acting as protein decoys”
(Chan and Pek 2019, doi:10.1016/j.tibs.2018.09.016).
-
Long noncoding RNAs
“Yao et al. review functions of lncRNAs in controlling chromatin
architecture, transcription and nuclear bodies in the nucleus and in
modulating mRNA stability, translation and protein modifications in the
cytoplasm”
(TOC blurb for Yao, Wang and Chen 2019, doi:10.1038/s41556-019-0311-8).
Summary of lncRNA functions. In nucleus:
- Regulate chromosome architecture.
- Modulate intra- and inter-chromosomal interactions.
- Promote or prevent recruitment of chromatin modifiers.
- Regulate transcription by forming R-loops
- Interfere with RNA polymerase II activity
- Directly regulate transcription via the lncRNA locus itself, or the transcription of this locus.
- Act as architectural agents in the formation of nuclear bodies.
- Participate in the functioning of nuclear bodies.
- Regulate mRNA turnover.
- Regulate translation.
- Modulate post-transcriptional modification of proteins.
In a screen of 16,401 lncRNA loci, 499 were found to be “required for robust
cellular growth”. And, of those, 89% affected growth in only one of the the
seven tested human cell lines. “Of note, not a single lncRNA, of 1,329
lncRNA genes tested, modified growth across all cell lines, suggesting that
lncRNA function is highly cell-type-specific”
(Koch 2017, doi:10.1038/nrg.2016.168, reporting on work by Liu et al. 2017,
doi:10.1126/science.aah7111). Given that there are not just seven, but
hundreds of human cell types, and given that the test was only for “robust
cellular growth” and not any of the countless other meaningful roles DNA
sequences can play, it seems a safe bet that the 499 loci are only the tip
of the iceberg.
“lncRNAs [long noncoding RNAs] fulfill regulatory roles at almost every
stage of gene expression, from targeting epigenetic modifications in the
nucleus to modulating mRNA stability and translation in the cytoplasm”.
“lncRNAs have, in a relatively short period of time, become recognized as a
legitimate and major new class of genes. lncRNAs may potentially comprise
a major component of the genome’s information content, complementary and
comparable in abundance and complexity to the proteome” (Mercer and Mattick
2013).
Referring to the “Wild West” landscape of transcription: “an average of 10
transcription units, the vast majority of which make long noncoding RNAs
(lncRNAs), may overlap each traditional coding gene. These lncRNAs include
not only antisense, intronic, and intergenic transcripts, but also
pseudogenes and retrotransposons” (Lee 2012).
lncRNAs “have, on average, a lower level of expression than protein coding
genes”. While their half-lives vary over a wide range and are comparable
to those of mRNAs,
they “appear to be more structured and stable than mRNA transcripts”. And
their expression is highly specific to cell type, “reflecting the
particular developmental stage and external environment that the cell has
experienced”. They tend to localize to the nucleus, but also (see below)
have important cytoplasmic functions (Batista and Chang 2013).
“[lncRNAs] regulate every process under the sun” (John Rinn, an RNA
researcher at Harvard Medical School, quoted in Saey 2011). There is
“emerging evidence that lncRNAs can assemble into ribonucleoprotein
complexes and contribute to gene regulation by mechanisms almost as diverse
as those employed by more conventional protein regulators (Conaway 2012).
“lncRNAs have no common mode of action and regulate gene expression through
comprehensive mechanisms such as chromatin remodeling, transcriptional
control, mRNA editing, splicing, and decay and control of protein synthesis.
Moreover, lncRNAs can act as guide molecules and protein scaffolds and
contribute to the formation of cellular substructures ... Several
cell-type-specific and abundant lncRNAs have been described as affecting the
maturation or activity of individual miRNAs through interactions with
miRNA-hosting transcripts or proteins involved in miRNA biogenesis ...
Alternatively, lncRNAs can act as endogenous sponges that titrate miRNAs and
inhibit their function” (Krol 2017, doi:10.1038/nsmb.3479).
“Approximately 10- to 20-fold more genomic sequence is transcribed to
lncRNA than to protein-coding RNA ... A rash of recent papers reveals that
lncRNAs are important and powerful cis- and trans-regulators
of gene activity that can function as scaffolds for chromatin-modifying
complexes and nuclear bodies, as enhancers and as mediators of long-range
chromatin interactions” (Nagano and Fraser 2011).
“In light of recent discoveries and given the diversity and flexibility of
long ncRNAs and their abilities to nucleate molecular complexes and to form
spatially compact arrays of complexes, it becomes likely that many or most
ncRNAs act as sensors and integrators of a wide variety of regulated
transcriptional responses and probably epigenetic events. ... We suggest
that a ncRNA/RNA-binding protein-based strategy, perhaps in concert with
several other mechanistic strategies, serves to integrate transcriptional,
as well as RNA-processing, regulatory programs” (Wang, Song, Glass and
Rosenfeld 2011).
“Both nuclear- and mitochondrial DNA-encoded lncRNAs mediate an intense
intercompartmental cross-talk, which opens a rich field for investigation of
the mechanism underlying the intercompartmental coordination and the
maintenance of whole cell homeostasis”
(Dong, Yoshitomi, Hu and Cui 2017, doi:10.1186/s13072-017-0149-x).
“An emerging concept is that lncRNAs serve as protein scaffolds, forming
ribonucleoproteins and bringing proteins in proximity ... We predicted the
largest human lncRNA–protein interaction network to date using the catRAPID
omics algorithm. In combination with tissue expression and statistical
approaches, we identified 847 lncRNAs (∼5% of the long non-coding
transcriptome) predicted to scaffold half of the known protein complexes and
network modules. Lastly, we show that the association of certain lncRNAs to
disease may involve their scaffolding ability. Overall, our results suggest
for the first time that RNA-mediated scaffolding of protein complexes and
modules may be a common mechanism in human cells”
(Ribeiro, Zanzoni, Cipriano et al. 2018, doi:10.1093/nar/gkx1169).
From a paper about the difficulties of lncRNA annotation:
“We must take care to focus efforts on collecting lncRNAs of biological
relevance. Unfortunately, we remain far from having reliable methods for
distinguishing functional lncRNAs from transcriptional noise. Although
imposing a minimum expression threshold is an obvious path, the discovery of
apparently functional lncRNAs with expression of <<1 copy per cell
would argue against imposing a hard expression cut-off. ... A question of
singular importance to the design of annotation projects is: is the lncRNA
population finite, and if so, how many transcripts and loci does it
comprise? Or conversely, is an effort at complete annotation doomed by the
fact that the transcriptome is infinite, owing to pervasive transcription or
unlimited combinatorial splicing? Certainly, after a decade of research, we
are little closer to assigning an upper bound to the first question. Recent
CLS studies finished sequencing before saturating even already known lncRNA
loci, while a recent study claims that lncRNA genes explore astronomical
numbers of available splicing combinations. Furthermore, present upper
estimates of lncRNA numbers are biased towards adult cell types, raising the
possibility of existence of untold numbers of developmentally regulated
lncRNAs.” “Although it has been argued, quite reasonably, that many lncRNAs
may represent non-functional noise, the growing number of clearly documented
counter-examples suggests that at least a substantial fraction of
transcripts is functional in the strictest sense of enhancing organismal
fitness.” (Uszczynska-Ratajczak, Lagarde, Frankish et al., 2018;
doi:10.1038/s41576-018-0017-y)
“The observation that long noncoding RNAs (lncRNAs) represent the majority
of transcripts in humans has led to a rapid increase in interest and study.
Most of this interest has focused on their roles in the nucleus. However,
increasing evidence is beginning to reveal even more functions outside the
nucleus, and even outside cells. Many of these roles are mediated by newly
discovered properties, including the ability of lncRNAs to interact with
lipids, membranes, and disordered protein domains, and to form
differentially soluble RNA–protein sub-organelles”. In particular:
“lncRNAs play important nucleating and structural roles in a growing number
of phase-separating ribonucleoprotein complexes; lncRNAs can interact with
membranes and specific phospholipids; lncRNAs can target proteins to
membranes; lncRNAs are important functional components of exosomes and are
likely to play roles in their formation and function”.
(Krause 2018, doi:10.1016/j.tig.2018.06.005)
“Evidence accumulated over the past decade shows that long non-coding RNAs
(lncRNAs) are widely expressed and have key roles in gene regulation. Recent
studies have begun to unravel how the biogenesis of lncRNAs is distinct from
that of mRNAs and is linked with their specific subcellular localizations
and functions. Depending on their localization and their specific
interactions with DNA, RNA and proteins, lncRNAs can modulate chromatin
function, regulate the assembly and function of membraneless nuclear bodies,
alter the stability and translation of cytoplasmic mRNAs and interfere with
signalling pathways. Many of these functions ultimately affect gene
expression in diverse biological and physiopathological contexts, such as in
neuronal disorders, immune responses and cancer. Tissue-specific and
condition-specific expression patterns suggest that lncRNAs are potential
biomarkers and provide a rationale to target them clinically”
(Statello, Guo, Chen and Huarte 2021, doi:10.1038/s41580-020-00315-9).
“Interestingly, a recent study has shown that lncRNAs with similar k-mer
content have related functions despite their lack of linear homology.
This study implies that short sequence elements in lncRNAs mediate
interactions with proteins (and/or other molecules), and thus are key
determinants of lncRNA function. However, the nature and dynamics of such
interactions still need to be elucidated. It is also increasingly evident
that multiple features of lncRNAs can define their functionality. These
features include their sequence, expression levels, processing, cellular
localization, structural organization and interactions with other molecules.
The integrated knowledge of all these features will hopefully increase the
identification and classification of functional lncRNAs”
(Statello, Guo, Chen and Huarte 2021, doi:10.1038/s41580-020-00315-9).
“Parental diet is known to influence the offspring in an intergenerational
manner, and this has been implicated in species adaptation and general
health. Recent studies highlight the role of maternal long noncoding RNAs
in serving as one of the 'memories' of maternal diet in regulating
offspring development and predisposition to metabolic disease”
(Koh and Pek 2023, doi:10.1016/j.tig.2022.07.006).
- Batut and Gingeras, working with five Drosophila species, investigated gene expression patterns during different stages of early development. They found 3973 promoters, mostly uannotated and widely associated with noncoding DNA, that drove expression during embryonic development. “We propose a hierarchical regulatory model in which core promoters define broad windows of opportunity for expression, by defining a range of transcription factors from which they can receive regulatory inputs. This two-tiered mechanism globally orchestrates developmental gene expression, including extremely widespread noncoding transcription. The sequence and expression specificity of noncoding RNA promoters are evolutionarily conserved, implying biological relevance. Overall, this work introduces a hierarchical model for developmental gene regulation, and reveals a major role for noncoding transcription in animal development. (Batut and Gingeras 2017, doi:10.7554/eLife.29005).
- “Attenuation of pre-rRNA synthesis in response to elevated temperature is accompanied by increased levels of PAPAS (“promoter and pre-rRNA antisense”), a long noncoding RNA (lncRNA) that is transcribed in an orientation antisense to pre-rRNA. Here we show that PAPAS interacts directly with DNA, forming a DNA–RNA triplex structure that tethers PAPAS to a stretch of purines within the enhancer region, thereby guiding associated CHD4/NuRD (nucleosome remodeling and deacetylation) to the rDNA promoter. ... The N-terminal part of CHD4 interacts with an unstructured A-rich region in PAPAS. ... Stress-dependent up-regulation of PAPAS is accompanied by dephosphorylation of CHD4 at three serine residues, which enhances the interaction of CHD4/NuRD with RNA and reinforces repression of rDNA transcription. The results emphasize the function of lncRNAs in guiding chromatin remodeling complexes to specific genomic loci and uncover a phosphorylation-dependent mechanism of CHD4/NuRD-mediated transcriptional regulation” (Zhao, Sentürk, Song and Grummt 2018, doi:10.1101/gad.311688.118).
-
Regulation of transcription initiation
- “The classical noncoding U1 snRNA, a component of the spliceosome, interacts with transcriptional initiation factor TFIIH to boost initiation rates of the basal transcriptional complex. Novel lncRNAs have demonstrated similar capabilities, bypassing chromatin-modifying complexes to communicate directly with gene promoters, the basal transcriptional machinery, and transcription factors. These lncRNAs are usually synthesized from regulatory loci such as enhancers and promoters and act in cis to mediate rapid, sensitive, and localized transciptional regulation” (Yang, Froberg and Lee 2014).
- “Recent studies have uncovered more lncRNAs that function as transcriptional activators in both mice and humans. Many of these ... help with the recruitment of protein factors to enhancers. ... The transcription of the noncoding transcripts at enhancers is also propsed to play a role in enhancer activation by mediating the deposition of H3K4 mono- and dimethylation” (Yang, Froberg and Lee 2014).
- The dihydrofolate reductase (DHFR) gene is repressed by a long noncoding RNA that is thought to “form a triplex with the major DHFR promoter and bind to TFIIB to displace the preinitiation complex from the DHFR locus, thereby blocking gene expression. ... Similarly, murine B2 RNA and human Alu RNA ... mediate repression of heat shock genes by binding to and deactivating RNAP II. Although these RNAs all bind the transcription initiation complex, they bear little resemblance to each other in sequence or structure” (Yang, Froberg and Lee 2014). This suggests something of the complex world of form and regulatory possibility presented by both the elements of the pre-initiation complex and long noncoding RNAs.
- “In fission yeast, glucose starvation triggers lncRNA transcription across promoter regions of stress-responsive genes ... Here, we demonstrate that such upstream noncoding transcription facilitates promoter association of the stress-responsive transcriptional activator Atf1 at the sites of transcription, leading to activation of the downstream stress genes ... These Atf1-binding sites exhibit low Atf1 occupancy and high histone density in glucose-rich conditions, and undergo dramatic changes in chromatin status after glucose depletion: enhanced Atf1 binding, histone eviction, and histone H3 acetylation. We also found that upstream transcripts bind to the Groucho-Tup1 type transcriptional corepressors Tup11 and Tup12, and locally antagonize their repressive functions on Atf1 binding. These results reveal a new mechanism in which upstream noncoding transcription locally magnifies the specific activation of stress-inducible genes via counteraction of corepressors” (Takemata, Oda, Yamada et al. 2016, doi:10.1093/nar/gkw142).
- “We used allele-sensitive single-cell RNA sequencing to demonstrate that, compared to messenger RNAs, lncRNAs have twice as long duration between two transcriptional bursts. Additionally, we observed increased cell-to-cell variability in lncRNA expression due to lower frequency bursting producing larger numbers of RNA molecules. Exploiting heterogeneity in asynchronously growing cells, we identified and experimentally validated lncRNAs with cell state-specific functions involved in cell cycle progression and apoptosis. Finally, we identified cis-functioning lncRNAs and showed that knockdown of these lncRNAs modulated the nearby protein-coding gene’s transcriptional burst frequency or size. In summary, we identified distinct transcriptional regulation of lncRNAs and demonstrated a role for lncRNAs in the regulation of mRNA transcriptional bursting” (Johnsson, Ziegenhain, Hartmanis et al. 2022, doi:10.1038/s41588-022-01014-1).
- See also this item under miRNAs can enhance gene expression.
-
Allele-specific roles
- Long noncoding RNAs (for example, the “classic” lncRNAs, XIST, Airn, and Kcnq1ot1 — and many others more recently discovered) play crucial repressive roles in allele-specific gene expression — for example, in X chromosome inactivation and gene imprinting. To take one case: “In mammalian imprinting, the noncoding RNA Air (also known as Airn) is expressed from the paternal chromosome and is involved in silencing the paternal alleles of multiple genes”. Repression of one of these genes is achieved by transcriptional interference: the promoter of that gene is overlapped by the lncRNA, so that expression of the latter blocks transcription of the imprinted gene (Batista and Chang 2013). Beyond imprinting: “Such a repressor function for lncRNA transcriptional overlap reveals a gene silencing mechanism that may be widespread in the mammalian genome, given the abundance of lncRNA transcripts” (Latos, Pauler, Koerner et al. 2012).
- Some lncRNAs have very short half-lives, and this seems to be important in cases of allele-specific regulation, as in X chromosome inactivation and imprinting (Lee 2012); that is, the lncRNA helps to establish a repressive condition at the site of transcription and is then degraded, preventing it from ectopically affecting other parts of the genome.
- “The fact that on the X-chromosome, as well as the imprinted loci, genes can escape from the silencing compartment into the transcriptionally active domains, despite the presence of the perpetrating lncRNA and repressive chromatin complexes in the vicinity, also suggests an additional layer of regulatory control that governs exit from the silencing compartment” (Saxena and Carninci 2011).
-
Role in epigenetic regulation
- Long noncoding RNAs “have been implicated in global remodeling of the epigenome and gene expression during reprogramming of somatic cells to induced pluripotent stem cells” (Nagano and Fraser 2011).
- “Many lncRNAs bind to chromatin-modifying proteins and recruit their catalytic activity to specific sites in the genome, thereby modulating chromatin states and impacting gene expression. Considering this regulatory potential in combination with the abundance of lncRNAs suggests that lncRNAs may be part of a broad epigenetic regulatory network” (Mercer and Mattick 2013).
- Example: “The lncRNA HOTAIR guides chromatin proteins and their catalytic action in trans to multiple sites spread across the genome. [The HOTAIR gene] is expressed from the end of the HOXC [gene] cluster in cells with distal and posterior identities. HOTAIR binds and targets PRC2 to the HOXD cluster as well as hundreds of additional sites around the genome to impart repressive histone modifications. These focal interactions of HOTAIR with target genome sites are likely pioneering events that subsequently nucleate broad regions of Polycomb occupancy and H3K27 trimethylation. By the expression of HOTAIR, distal developmental states can initiate an epigenetic regulatory cascade that maintains the cells’ positional identity and continually refines a progressive developmental trajectory” (Mercer and Mattick 2013).
- The act of transcribing a long noncoding RNA can itself result in the establishment of a repressive chromatin state in adjacent DNA regulatory elements — for example, a gene promoter. More generally, transcription of lncRNAs “can influence gene expression and genome organization by promoting chromatin modifications, by recruiting gene active regions to common transcription factories, or by exposing the DNA strands to enzymatic activity. Hence the presence of multiple lncRNA genes in a region may help chromosomal loci adopt distinct conformation with transcriptional activation. For example, in the Hox loci, collinear expression of Hox mRNA genes and Hox lncRNAs along the chromosome is associated with the progressive recruitment of those chromosomal segments into a tightly interacting domain that is distinct from the transcriptionally silent portion of the loci” (Batista and Chang 2013).
- This effect of transcription upon neighboring loci can be combined with the means to regulate genes from afar. For example, transcription of the Airn lncRNA inhibits the Igfr2 gene, but Airn then targets a protein and a histone tail modification to silence other, more distant genes on the paternal chromosome (Batista and Chang 2013).
- “we identify new fission yeast regulatory lncRNAs that are targeted, at their site of transcription, by the YTH domain of the RNA‐binding protein Mmi1 and degraded by the nuclear exosome. We uncover that one of them, nam1, regulates entry into sexual differentiation. Importantly, we demonstrate that Mmi1 binding to this lncRNA not only triggers its degradation but also mediates its transcription termination, thus preventing lncRNA transcription from invading and repressing the downstream gene encoding a mitogen‐activated protein kinase kinase kinase (MAPKKK) essential to sexual differentiation. In addition, we show that Mmi1‐mediated termination of lncRNA transcription also takes place at pericentromeric regions where it contributes to heterochromatin gene silencing together with RNA interference (RNAi). These findings reveal an important role for selective termination of lncRNA transcription in both euchromatic and heterochromatic lncRNA‐based gene silencing processes” (Touat‐Todeschini, Shichino, Dangin et al. 2017, doi:10.15252/embj.201796571).
- lncRNAs can be cleaved to generate small RNAs. For example, “the formation of extended RNA duplexes or stem loops provides a ready substrate for Dicer enzyme to generate multiple small regulatory RNAs that have cascading ability to mediate downstream epigenetic changes. ... Comparison between long and short RNA populations in human cells suggests widespread evidence of post-transcriptional cleavage, with lncRNAs being a preferred substrate for the generation of small RNAs”. Thus, it begins to look as though RNAs in general constitute “a standard medium for transferring information within and between regulatory pathways, thereby assembling complex, multilayered and modular regulatory networks in the cell” (Mercer and Mattick 2013).
-
Enhancer-like functions
- A survey of more than a thousand long noncoding RNAs produced a set of them that acted like enhancers. That is, they activated expression of protein-coding genes located near to the DNA sequences producing the RNAs. The method of activation is not known. (Ørom, Derrien, Beringer et al. 2010; Ørom and Shiekhattar 2011; Ørom and Shiekhattar 2011).
- One example: in vertebrates a DNA site called HOTTIP is brought by chromosome looping into proximity to a set of Hox genes. The long noncoding RNA produced from HOTTIP then associates with certain adaptor proteins that restructure the local chromatin in order to coordinate gene activation (Wang, Yang, Liu et al. 2011).
- “The GAL gene cluster of the yeast Saccharomyces cerevisiae encodes a series of three inducible genes that are turned on or off by the presence or absence of specific carbon sources in the environment. Previous studies have documented the presence of two lncRNAs—GAL10 and GAL10s—encoded by genes that overlap the GAL cluster. We have now uncovered a role for both these lncRNAs in promoting the activation of the GAL genes when they are released from repressive conditions. This activation occurs at the kinetic level, through more rapid recruitment of RNA polymerase II and decreased association of the co-repressor, Cyc8. Under normal conditions, but also especially when they are stabilized and their levels are up-regulated, these GAL lncRNAs promote faster GAL gene activation. We suggest that these lncRNA molecules poise inducible genes for quick response to extracellular cues, triggering a faster switch in transcriptional program” (Cloutier, Wang, Ma et al. 2013).
- “We characterize a new class of lncRNAs called super-lncRNAs that target super-enhancers and which can contribute to the local chromatin organization of the super-enhancers ... we identify 442 unique super-lncRNA transcripts in 27 different human cell and tissue types; 70% of these super-lncRNAs were tissue restricted. They primarily harbor a single triplex-forming repeat domain, which forms an RNA:DNA:DNA triplex with multiple anchor DNA sites (originating from transposable elements) within the super-enhancers. Super-lncRNAs can be grouped into 17 different clusters based on the tissue or cell lines they target. Super-lncRNAs in a particular cluster share common short structural motifs and their corresponding super-enhancer targets are associated with gene ontology terms pertaining to the tissue or cell line. Super-lncRNAs may use these structural motifs to recruit and transport necessary regulators (such as transcription factors and Mediator complexes) to super-enhancers, influence chromatin organization, and act as spatial amplifiers for key tissue-specific genes associated with super-enhancers” (Soibam 2017, doi:10.1261/rna.061317.117).
-
Co- and post-transcriptional regulation by lncRNAs
- “Co- and post-transcriptional processes such as splicing, transport, translation of mRNA, and subcellular localization of proteins may also be controlled by lncRNAs. Interaction of lncRNAs with primary coding transcripts can occlude splice junctions and result in production of alternative isoforms (Yang, Froberg and Lee 2014).
- The Zeb2 transcription factor is implicated in the epithelial-mesenchymal transition during embryogenesis and cancer transformation. Zeb2 is regulated post-transcriptionally by its natural antisense transcript (a long noncoding RNA), synthesized from the antisense strand of the Zeb2 promoter. This lncRNA “shields an internal ribosome entry site within the 5' untranslated region of Zeb2 from mRNA splcing, thereby allowing for increased rates of Zeb2 translation and driving the epithelial-mesenchymal transition” (Yang, Froberg and Lee 2014).
- A long noncoding RNA is transcribed antisense to Uchl1, a gene implicated in brain function and neurodegenerative diseases such as Parkinson’s and Alzheimer’s. Under conditions of stress, the antisense lncRNA — which partially overlaps with the 5' end of Uchl1 — is exported to the cytoplasm. There it enhances translation of Uchl1 by recruiting ribosomes to the mRNA. The overlap is crucial for lncRNA function.
- Long noncoding RNAs can perform a role similar to mRNAs in acting as “decoys” for miRNAs, thus competing with the mRNA targets of those miRNAs. By this means, for example, a long noncoding RNA “plays an important role in muscle differentiation”. It does so by “soaking up” miRNAs that otherwise would target two transcription factors that activate muscle-specific gene expression (Cesana, Cacchiarelli, Legnini et al. 2011).
- Another way long noncoding RNAs can act as “decoys”: because they reproduce the “genetic code” of the DNA section from which they derive, they can serve as alternative targets for DNA-binding regulatory proteins, thereby depriving DNA of those proteins (Rinn and Chang 2012). In other words, this is one of the ways the cell can “regulate the regulators”
- One research group has shown that a splice site in a long noncoding RNA can be strongly regulative of transcription of a neighboring gene, reducing transcription by 94%. More generally, the promoters and transcription of lncRNAs can both play gene-regulatory roles via multiple regulatory paths. And “because there exist thousands of other loci that fit our selection criteria, we expect that similar mechanisms broadly contribute to gene regulation in many loci” (Engreitz, Haines, Perez et al. 2016, doi:10.1038/nature20149).
-
Interaction with proteins
- Overall, at least 15% of proteins are associated with polyadenylated RNA (Mercer and Mattick 2013). (lncRNAs, unlike small RNAs, are often polyadenylated.)
- “It is clear that many chromatin regulatory complexes moonlight as RNA-binding proteins; the ability to bind lncRNAs endows them with condition- or allele-specific recognition of target gene chromatin”. Acting as guides, lncRNAs “combine two basic molecular functions — binding of a protein partner plus a mechanism to interface with selective regions of the genome” (Rinn and Chang 2012).
- “With at least 12 chromatin-modifying proteins having been associated with lncRNAs to date, the composition of possible chromatin-modifying proteins in a single ribonucleoprotein can be varied by shuffling the modular components within an lncRNA ... For example, in mice the lncRNA Kcnq1ot1 can recruit both PRC2 and G9a, which impart [histone tail modifications with opposite effect] H3K4 trimethylation and H3K9 methylation, respectively” (Mercer and Mattick 2013).
- The secondary folding structure of a long noncoding RNA is often central to its functioning. For example, the function of the tumor suppressor lncRNA, MEG3, has been evolutionarily conserved by means of the conservation of its structure, not its sequence (Mercer and Mattick 2013).
- “Proteins tend to interact with RNA where it forms complex secondary structures ... Almost all such interactions characterized to date involve conformational changes to the protein, the RNA or both”. Such conformational changes can help determine what other proteins can join the molecular complex, leading to many different combinatorial possibilities. (Mercer and Mattick 2013).
- A new role for long noncoding RNA in gene regulation has now been found. The demonstration case involves X chromosome inactivation, where the CTCF protein sits on the promoter of a key gene involved in X chromosome inactivation, preventing the gene from being expressed. However, when it is time for inactivation (that is, time for the expression of the repressed gene), a long noncoding RNA binds to CTCF and lifts it from the promoter. There appears to be a lot of complexity, not yet unravelled, concerning how the noncoding RNA distinguishes the instances of CTCF on the relevant X chromosome gene from all the many other instances of CTCF bound to DNA — and also about how the correct timing is established. Many other factors are involved in X chromosome inactivation (Sun, Del Rosario, Szanto et al. 2013).
- “Pumilio homologue 1 (PUM1) and PUM2 are RNA-binding proteins that bind to motifs known as pumilio response elements in many mRNAs and stimulate their degradation. The levels of PUM proteins must be strictly controlled to avoid pathologies such as neurodegeneration, but how this is achieved is unknown. Lee et al. identified a long non-coding RNA, which they termed NORAD (non-coding RNA activated by DNA damage), and showed that it sequesters PUM proteins and suppresses PUM-mediated RNA degradation and genomic instability” (Zlotorynski 2016, doi:10.1038/nrm.2016.5).
- “The lncRNA NEAT1 uses miRNA-mimicking features to anchor the Microprocessor complex to nuclear subdomains called paraspeckles. The paraspeckle-associating proteins NONO and PSF then interact with NEAT1 and different pri-miRNAs to regulate pri-miRNA processing and form the nuclear lncRNA-organized substructure that influences global miRNA biogenesis” (Krol 2017, doi:10.1038/nsmb.3479).
- More broadly about paraspeckles: “Nascent 23 000-nucleotide NEAT1 long noncoding RNA transcripts act as a seed to recruit nuclear RNA-binding proteins and build a paraspeckle. Protein domains that mediate liquid-liquid phase separation are essential for many aspects of paraspeckle formation, including gluing together individual ribonucleoprotein bundles into a mature paraspeckle. Paraspeckle formation is dynamic and triggered by many different cell stress scenarios including infection and transformation. New discoveries show how paraspeckles are formed through multiple RNA-protein and protein-protein interactions, some of which involve extensive polymerization, and others with multivalent interactions driving phase separation. Once formed, paraspeckles influence gene regulation through sequestration of component proteins and RNAs, with subsequent depletion in other compartments. [We find today] an emerging role for these dynamic bodies in a multitude of cellular settings” (Fox, Nakagawa, Hirose and Bond 2018, doi:10.1016/j.tibs.2017.12.001).
- See also under “Competing endogenous RNAs” above.
-
Interaction with other noncoding RNAs
- “RNase P-mediated endonucleolytic cleavage plays a crucial role in the 3' end processing and cellular accumulation of MALAT1, a nuclear-retained long noncoding RNA that promotes malignancy ... Here we characterize a broadly expressed natural antisense transcript at the MALAT1 locus, designated as TALAM1, that positively regulates MALAT1 levels by promoting the 3' end cleavage and maturation of MALAT1 RNA. TALAM1 RNA preferentially localizes at the site of transcription, and also interacts with MALAT1 RNA. Depletion of TALAM1 leads to defects in the 3' end cleavage reaction and compromises cellular accumulation of MALAT1. Conversely, overexpression of TALAM1 facilitates the cleavage reaction in trans. Interestingly, TALAM1 is also positively regulated by MALAT1 at the level of both transcription and RNA stability. Together, our data demonstrate a novel feed-forward positive regulatory loop that is established to maintain the high cellular levels of MALAT1, and also unravel the existence of sense-antisense mediated regulatory mechanism for cellular lncRNAs that display RNase P-mediated 3' end processing” (Zong, Nakagawa, Freier et al. 2016, doi:10.1093/nar/gkw047).
-
Role in nuclear organization
“RNA, DNA, and protein molecules are highly organized within
three-dimensional (3D) structures in the nucleus. Although RNA has been
proposed to play a role in nuclear organization, exploring this has been
challenging because existing methods cannot measure higher-order RNA and
DNA contacts within 3D structures. To address this, we developed RNA &
DNA SPRITE (RD-SPRITE) to comprehensively map the spatial organization of
RNA and DNA. These maps reveal higher-order RNA-chromatin structures
associated with three major classes of nuclear function: RNA processing,
heterochromatin assembly, and gene regulation. These data demonstrate
that hundreds of ncRNAs form high-concentration territories throughout
the nucleus, that specific RNAs are required to recruit various
regulators into these territories, and that these RNAs can shape
long-range DNA contacts, heterochromatin assembly, and gene expression.
These results demonstrate a mechanism where RNAs form high-concentration
territories, bind to diffusible regulators, and guide them into
compartments to regulate essential nuclear functions”
(Quinodoz, Jachowicz, Bhat, Thai and Plath 2021,
doi:10.1016/j.cell.2021.10.014).
- Nascent noncoding RNAs “can trigger assembly of various nuclear bodies by serving as scaffolds for accumulation of specific proteins”. For example, paraspeckles, implicated in the regulation of hyper-edited mRNAs, are assembled on certain long noncoding RNAs as they are being transcribed (Nagano and Fraser 2011).
- An example involving long noncoding RNAs, a chromatin remodeling protein, post-translational modification of the protein, spatial organization of the nucleus, and gene expression: the protein, PRC2 (polycomb repressor complex 2), when methylated, represses a group of genes. It does so through its association with a long noncoding RNA, as a result of which the genes are located in repressive compartments of the nucleus known as Polycomb Group (PcG) bodies. But when PRC2 is demethylated, it associates with a different long noncoding RNA, and this relocates the genes to interchromatin granules (Yang, Lin, Liu et al. 2011; also Batista and Chang 2013).
- Another example, rather complex, but complexity is really the byword in all this: a certain imprinted region of the genome connected with Prader-Willi syndrome “hosts multiple intron-derived lncRNAs with small nucleolar RNAs at their ends — so called ‘sno-lncRNAs.’ It is probable that the presence of structured snoRNAs at the ends of lncRNAs stabilizes these molecules, which have no 5' cap or polyA tail. These RNAs are retained in the nucleus and localize to, or remain near, their sites of transcription. Knockdown of sno-lncRNAs has little effect on the expression of nearby genes, suggesting that it does not affect gene expression in cis. Instead, these sno-lncRNAs seem to create a [nuclear] ‘domain’ where the splicing factor Fox2 is enriched. These sno-lncRNAs contain multiple binding sites for Fox2, and altering the level of sno-lncRNAs led to a redistribution of Fox2 in the nucleus and changes in mRNA splicing patterns. Hence, the sno-lncRNAs appear to function as Fox2 sinks, participating in the regulation of splicing in specific subnuclear domains” (Batista and Chang 2013).
- lncRNAs can not only cooperate in bringing molecular “communities” together, but can also keep them apart until the proper time. “For example, certain environmental stresses trigger the retention of select proteins in the nucleolous away from their normal site of action. The retention at the nucleolus requires a signal sequence and the expression of specific noncoding RNAs expressed from the large intergenic spacer [IGS] of the rDNA repeats. ... Unique IGS ncRNAs are transcriptionally induced by specific stressors, functioning as baits for proteins with specific signal sequences” (Batista and Chang 2013).
- In a rather startling finding, it’s been shown that the lncRNA XIST, a key player in X chromosome inactivation, “operates by interacting with loops of nearby chromosome. ‘It seems to be creating a three-dimensional organization, bringing together regions of the genome in a way that we had assumed proteins were doing’, says Emmanouil Dermitzakis ... This finding supports a role for lncRNAs in regulating chromosomal activity by influencing the shape of chromatin ... Preliminary results with other lncRNAs suggest that they, too, may work like XIST” (Pennisi 2013; Engreitz, Pandya-Jones, McDonel et al. 2013).
- One lncRNA has now been found that apparently works across multiple chromosomes: “We describe one lncRNA, Firre, that interacts with the nuclear-matrix factor hnRNPU through a 156-bp repeating sequence and localizes across an ~5-Mb domain on the X chromosome. We further observed Firre localization across five distinct trans-chromosomal loci, which reside in spatial proximity to the Firre genomic locus on the X chromosome. Both genetic deletion of the Firre locus and knockdown of hnRNPU resulted in loss of colocalization of these trans-chromosomal interacting loci. Thus, our data suggest a model in which lncRNAs such as Firre can interface with and modulate nuclear architecture across chromosomes” (Hacisuleyman, Goff, Trapnell et al. 2014).
- “In a recent study, Vilborg et al. describe a new class of long chromatin-associated RNAs that are generated by readthrough transcription and are highly inducible by osmotic stress. The authors discovered a long non-coding RNA that was upregulated in response to osmotic stress induced by treatment with KCl, NaCl or sucrose ... sequence mapping revealed its inclusion within a transcript generated by readthrough transcription from the gene upstream. This is termed a 'downstream of gene'-containing transcript (DoG). Remarkably, bioinformatic analysis identified KCl-induced DoGs downstream of more than 10% of all human protein-coding genes, suggesting a global effect of osmotic stress on transcription downstream of genes ... induction of DoGs by KCl is mediated by increased readthrough rather than upregulation of the upstream gene ... PASs [polyadenylation signals] were depleted in DoG-associated genes, which should decrease the efficiency of transcription termination, providing an explanation for increased readthrough in DoG-associated genes ... the authors hypothesized a role for DoGs in reinforcing chromatin against nuclear shrinkage caused by osmotic stress. Indeed ... KCl treatment caused chromatin collapse and holes in nuclei that were more severe in cells that had been pretreated with an inhibitor ... to prevent DoG induction” (Waldron 2015, doi:10.1038/nrg3994).
- “Paraspeckles are nuclear condensates, or membranelees organelles, that are built on the long noncoding RNA, NEAT1, and have been linked to many diseases. Although originally described as constitutive structures, here, in reviewing this field, we develop the hypothesis that cells increase paraspeckle abundance as part of a general stress response, to aid pro‐survival pathways. Paraspeckles increase in many scenarios: when cells transform from one state to another, become infected with viruses and bacteria, begin to degenerate, under inflammation, in aging, and in cancer. Cells increase paraspeckles by increasing transcription of NEAT1 and adjusting its RNA processing. These increases in NEAT1 are driven by numerous stress‐sensing signaling pathways, including signaling to mitochondria and stress granules, revealing crosstalk between the cytoplasm and nucleoplasm in the stress response. Thus, paraspeckles are an important piece of the puzzle in cellular homeostasis, and could be considered RNA‐scaffolded nuclear equivalents of dynamic stress‐induced structures that form in the cytoplasm. We speculate that, in general, cells rely on phase‐separated paraspeckles to transiently tweak gene regulation in times of cellular flux” (McCluggage and Fox 2021, doi:10.1002/bies.202000245).
-
Role in genomic stability
-
The human NORAD lncRNA “is regulated in response to DNA damage and
plays a key role in maintaining genome integrity by modulating the
activity the RNA binding proteins PUM2 and PUM1”. The deletion of
both NORAD alleles results in “a marked chromosomal instability,
characterized by a tendency to lose and gain chromosomes and an
increased frequency of spontaneous tetraploidization ... The NORAD RNA
is found almost exclusively in the cytoplasm, [and] multiple lines of
evidence indicate that NORAD affects genomic stability through its
direct interaction with Pumilio2 (PUM2) and possibly Pumilio 1 (PUM1),
two RNA-binding proteins [that] bind to the 3'UTR of target mRNAs via
an 8-nucleotide specific sequence and reduce their stability”.
“NORAD can bind to PUM2 with great affinity as a result of the presence of 15 Pumilio-binding motifs ... Given the high abundance of NORAD, and the presence of multiple binding sites on each transcript, Lee and colleagues propose that NORAD functions as a PUM2/PUM1 decoy, preventing these RNA-binding proteins from interacting with and destabilizing their targets ... If these results convincingly establish the functional relevance of the NORAD/Pumilio interaction, what remains unclear are the molecular mechanisms through which loss of this interaction leads to chromosomal instability. As noted by Lee and colleagues, several of the PUM2 targets that are downregulated upon NORAD inactivation are known to control genomic stability ...
“NORAD could exert some of its functions through additional mechanisms; and it is possible that the interaction with PUM1/2 is not exclusively inhibitory, but could rather modulate the activity of Pumilio (and NORAD). This is especially relevant because although Lee and colleagues found a significant enrichment for PUMILIO targets among the genes downregulated in NORAD–/– cells, the correlation was not absolute, with a large number of genes regulated by NORAD not being PUMILIO targets and vice versa” (Ventura 2016, doi:10.1016/j.tig.2016.04.002).
-
The human NORAD lncRNA “is regulated in response to DNA damage and
plays a key role in maintaining genome integrity by modulating the
activity the RNA binding proteins PUM2 and PUM1”. The deletion of
both NORAD alleles results in “a marked chromosomal instability,
characterized by a tendency to lose and gain chromosomes and an
increased frequency of spontaneous tetraploidization ... The NORAD RNA
is found almost exclusively in the cytoplasm, [and] multiple lines of
evidence indicate that NORAD affects genomic stability through its
direct interaction with Pumilio2 (PUM2) and possibly Pumilio 1 (PUM1),
two RNA-binding proteins [that] bind to the 3'UTR of target mRNAs via
an 8-nucleotide specific sequence and reduce their stability”.
-
Cytoplasmic functions
- “A substantial proportion of lncRNAs reside within, or are dynamically shuttled, to the cytoplasm where they regulate protein localization, mRNA translation and stability” (Mercer and Mattick 2013).
- “lncRNAs can ... ‘identify’ mRNAs in the cytoplasm and modulate their life cycle. Recent works demonstrated that lncRNAs impact both the mRNA half-life and translation of mRNAs”. For example, some lncRNAs interact with the Stau1 protein to promote the stability of mRNAs — the exact opposite of the destabilizing role of miRNAs and siRNAs. Other lncRNAs work with Stau1 to facilitate degradation of mRNAs (Batista and Chang 2013; see “Staufen1-mediated RNA decay” above.)
- There are other pathways for both repression and promotion of translation, involving antisense long noncoding RNAs (Batista and Chang 2013). For example, “By virtue of their ability to base pair with mRNAs, cytoplasmic lncRNAs also can regulate translation. The UCHL1 mRNA is complemented by an antisense lncRNA, which, in response to stress or the mTOR pathway, is shuttled to the cytoplasm where, via an antisense complementary to the UCHL1 AUG initiation codon and combined inverted SINEB2 domains, increases UCHL1 protein synthesis”. “More than half of mammalian coding genes have complementary noncoding antisense transcription”, much of which yields lncRNAs that can recognize the associated coding mRNAs in the cytoplasm. (Mercer and Mattick 2013).
- lncRNAs can guide the localization of cytoplasmic proteins. Example: “The NFAT transcription factor is trafficked from the cytoplasm to the nucleus to activate target genes in response to calcium-dependent signals. A lncRNA, NRON, complexes with importin-β proteins and regulates the trafficking of NFAT. Notably, NRON inhibits the trafficing of NFAT to the nucleus specifically, with other proteins also trafficked by importin-β proteins, such as NF-κB, being unaffected” (Mercer and Mattick 2013).
- “Recent evidence indicates that non‐coding RNAs (ncRNAs) may contribute to the synchronization of a series of essential cellular and mitochondrial biological processes, acting as “messengers” between the nucleus and the mitochondria. Here, we discuss the emerging putative roles of ncRNAs in various bidirectional signaling pathways established between the host cell and its mitochondria, and how the dysregulation of these pathways may lead to aging‐related diseases, including cancer, and offer new promising therapeutic avenues” (Vendramin, Marine and Leucci 2017, doi:10.15252/embj.201695546).
-
Partial translation of long noncoding RNAs
By definition, noncoding RNAs are noncoding, and hence should
not be translated. But it has been found that “many putative lincRNAs
[long noncoding RNAs] have successive short segments that are
translated at a rate similar to comparable classical protein coding
sequences” (Weiss and Atkins 2011). The functions of these segments
are only now beginning to be explored.
- “We discovered a conserved micropeptide, which we named myoregulin (MLN), encoded by a skeletal muscle-specific RNA annotated as a putative long noncoding RNA. MLN shares structural and functional similarity with phospholamban (PLN) and sarcolipin (SLN), which inhibit SERCA, the membrane pump that controls muscle relaxation by regulating Ca2+ uptake into the sarcoplasmic reticulum (SR). MLN interacts directly with SERCA and impedes Ca2+ uptake into the SR. In contrast to PLN and SLN, which are expressed in cardiac and slow skeletal muscle in mice, MLN is robustly expressed in all skeletal muscle. Genetic deletion of MLN in mice enhances Ca2+ handling in skeletal muscle and improves exercise performance. These findings identify MLN as an important regulator of skeletal muscle physiology and highlight the possibility that additional micropeptides are encoded in the many RNAs currently annotated as noncoding” (Anderson, Anderson, Chang et al. 2015, doi:10.1016/j.cell.2015.01.009).
- Protein coding DNA sequences (“exons”) within genes have distinctive compositional patterns. A team of researchers looked for the same patterns within lncRNAs — and found them, although they were somewhat less accentuated. “Specifically, compared with [lncRNA] introns, lncRNA exons are GC rich. Additionally we report evidence for the action of purifying selection to preserve exonic splicing enhancers within human multiexonic lncRNAs and nucleotide composition in fruit fly lncRNAs. Our findings provide evidence for selection for more efficient rates of transcription and splicing within lncRNA loci. Despite only a minor proportion of their RNA bases being constrained, multiexonic intergenic lncRNAs appear to require accurate splicing of their exons to transact their function” (Haerty and Ponting 2015, doi:10.1261/rna.047324.114).
- “Recent studies in flies and mammals have revealed that transcripts annotated as lncRNAs encode smORF [small open reading frame] peptides that bind to, and inhibit, the sarco/endoplasmic reticulum calcium adenosine triphosphatase (SERCA), an ion pump that is a key player in handling calcium in striated muscles ... Nelson et al. report that a lncRNA-encoded small peptide competes with SERCA-inhibitory peptides, thereby favoring heart contractility in mammals. These findings open new ways to understand cardiac function and pathologies, and show that smORF peptides act as versatile regulators of protein activity” (Payre and Desplan 2016, doi:10.1126/science.aad9873).
-
Role in development and disease
The expression of lncRNAs has been ... generally found to be more cell
type specific than the expression of protein-coding genes.
Interestingly, in several cases, such tissue specificity has been
attributed to the presence of transposable elements that are embedded
in the vicinity of lncRNA transcription start sites. Moreover, lncRNAs
have been shown to be differentially expressed across various stages of
differentiation, which indicates that they may be novel ‘fine-tuners’
of cell fate. This specific spatiotemporal expression can be linked to
the establishment of both well-defined barriers of gene expression and
cell-type-specific gene regulatory programmes. Combined with the
involvement of lncRNAs in positive or negative feedback loops, lncRNAs
can amplify and consolidate the molecular differences between cell
types that are required to control cell identity and lineage
commitment” (Fatica and Bozzoni 2014).
Some long noncoding RNAs are associated with induced pluripotency and
maintenance of embryonic stem cells.
- “At least 26 different lincRNAs [long intervening (or intergenic) noncoding RNAs] need to be on to keep an embryonic stem cell a stem cell. ... As stem cells transform into various types of cells, they turn off some specific lincRNAs and turn on others creating, a mix of activity that can define the cell” (Saey 2011).
- “Long noncoding RNAs (lncRNAs) regulate diverse processes, yet a potential role for lncRNAs in maintaining the undifferentiated state in somatic tissue progenitor cells remains uncharacterized...We identified ANCR (anti-differentiation ncRNA) as an 855-base-pair lncRNA [in humans] down-regulated during differentiation. Depleting ANCR in progenitor-containing populations, without any other stimuli, led to rapid differentiation gene induction...The ANCR lncRNA is thus required to enforce the undifferentiated cell state within epidermis”. There are, however, much wider effects of this lncRNA, with the expression of many genes throughout the genome being affected by it (Kretz, Webster, Flockhart et al. 2012).
- “Recent progress suggests that the involvement of lncRNAs in human diseases could be far more prevalent than previously appreciated” (Wapinski and Chang 2011).
- “Xist RNA has now been directly implicated in human cancers. Xist maintains dosage compensation for ~1000 genes on the X chromosome, several of which are putative oncogenes, thus, it is possible that misregulation of Xist contributes to cancer phenotypes through aberrations in expression of X-linked oncogenes. ... Direct causality has now emerged from an in vivo study in which deletion of Xist in the hematopoietic lineage resulted in the development of leukemia in mice with full penetrance. Gene expression profiling over the course of disease progression revealed significant upregulation of X-linked genes, suggesting the possibility of X reactivation following Xist loss” (Yang, Froberg and Lee 2014).
- “Overwhelming evidence reveals that large noncoding RNAs are molecules that keep in perfect tune the balance of gene expression networks, and discordance in their function results in homeostatic imbalance, ultimately causing cellular transformation [as in cancer]. Large ncRNAs are shedding new light on our understanding of these cancer pathways and may represent a ‘missing link’ in cancer” (Huarte and Rinn 2010).
- The tumor suppressor p53 regulates a number of long noncoding RNAs. Knocking out one of those RNAs (called lincRNA-p21) results in changed expression (mostly derepression) of more than 1000 genes (Nagano and Fraser 2011).
- “The interactions between long intergenic non-coding RNAs (lincRNAs) and proteins have roles in various cellular processes. By contrast, functional interactions of RNAs with phospholipids have yet to be identified. Lin et al. now report that lincRNA for kinase activation (LINK-A; also known as LINC01139) specifically interacts with the plasma membrane-associated lipid phosphatidylinositol-3,4,5-trisphosphate (PIP3) and with its effector protein AKT (also known as protein kinase B). These interactions activate AKT and promote tumorigenesis and resistance to AKT inhibitors” (Zlotorynski 2017, doi:10.1038/nrm.2017.18).
- In a number of distantly related butterfly species whose wing colors had been thought to be determined by a protein, it has now been established that a long noncoding RNA is the decisive regulator. This RNA is broken down into an miRNA that is the immediate regulator of color. “‘A lot is happening within this small part of the genome,’ says Violaine Llaurens, an evolutionary biologist at the College of France. She cautions that other regulatory elements probably play a role in butterfly wing patterns. But the fact that the same microRNA fine-tunes coloration in very distantly related species is ‘amazing,’ says Anyi Mazo-Vargas, an evolutionary biologist at Duke ... She suspects these RNAs color wings in most, if not all, of the 180,000 species of moths and butterflies” (Pennisi 2024, https://science.org/content/article/surprise-rnas-solve-mystery-how-butterfly-wings-get-their-colorful-patterns)
- Summary statement. “Genome-wide analyses have shown that virtually the entire genome is differentially transcribed in highly complex cell-specific patterns, to produce tens if not hundreds of thousands of long non-coding RNAs (lncRNAs). ... These lncRNAs are specifically expressed, especially in the brain, and are dynamically regulated during cell differentiation, including during embryonal and neural stem cell differentiation. ... A set of lncRNAs ... are dynamically regulated during the differentiation of human stem cells into neurons and ... are involved either in the maintenance of pluripotency or in neuronal differentiation. ... lncRNAs are also involved in embryonal stem cell maintenance and lineage specification. ... lncRNAs are associated with chromatin-modifying complexes, such as polycomb components and histone methyltransferases, extending earlier studies showing association of lncRNAs with both activating and repressive chromatin-modifying enzymes and states. The inescapable conclusion is that these RNAs are likely acting as adaptors to assemble different suites of generic effector proteins that are recognized and bound by secondary structural features embedded within the RNA, and to direct these to specific genomic positions by virtue of RNA–DNA interactions. This previously hidden world of RNA-directed epigenetic control of gene structure and expression may be extremely sophisticated, not simply operating at the regional level, but extending to individual exons and other features such as promoters and enhancers. ... Epigenetic control of splicing [has] obtained experimental support. ... Many if not most lncRNAs are themselves alternatively spliced, adding further complexity to this scenario. ... Clearly long-held ideas of gene regulation in development and cognition will have to be reassessed...” (Mattick 2012).
-
Repeats and repeat-derived RNAs
“Recent studies recognize a vast diversity of noncoding RNAs with largely
unknown functions, but few have examined interspersed repeat sequences,
which constitute almost half our genome”. It turns out that there is
“surprisingly abundant euchromatin-associated RNA comprised predominantly of
repeat sequences”, including LINE-1. This RNA, which is more abundant,
even, than ribosomal RNA (rRNA), is excluded from heterochromatin and
“strictly localizes to the interphase chromosome territory in cis and
remains stably associated with the chromosome territory following prolonged
transcriptional inhibition. The [repeat-derived] RNA territory resists
mechanical disruption and fractionates with the nonchromatin scaffold but
can be experimentally released. Loss of repeat-rich, stable nuclear RNAs
from euchromatin corresponds to aberrant chromatin distribution and
condensation ... These findings impact two “black boxes” of genome science:
the poorly understood diversity of noncoding RNA and the unexplained
abundance of repetitive elements (Hall, Carone, Gomez et al. 2014,
doi:10.1016/j.cell.2014.01.042).
Commenting on the study by Hall, Carone, Gomez et al. (above), another
researcher writes: “In addition to visualizing the life cycle of these
abundant and important repeats RNAs in association with chromatin, this
study helped me understand our own unexpected discovery that histones and
chromatin precipitate when RNA is removed. I realized that, particularly in
light of RNA abundance in chromatin, the negative charge of RNA might
influence the local ionic environment. This insight led to our recent
discovery that LINE1 RNAs bind to histones and can open compacted chromatin
by inhibiting electrostatic interactions between histones and DNA”
(Farnebo 2020, doi:10.1038/s41580-019-0200-9).
- “Transposable elements represent nearly half of mammalian genomes and are generally described as parasites, or ‘junk DNA.’ The LINE1 retrotransposon is the most abundant class and is thought to be deleterious for cells, yet it is paradoxically highly expressed during early development. Here, we report that LINE1 plays essential roles in mouse embryonic stem cells (ESCs) and pre-implantation embryos. In ESCs, LINE1 acts as a nuclear RNA scaffold that recruits Nucleolin and Kap1/Trim28 to repress Dux, the master activator of a transcriptional program specific to the 2-cell embryo. In parallel, LINE1 RNA mediates binding of Nucleolin and Kap1 to rDNA, promoting rRNA synthesis and ESC self-renewal. In embryos, LINE1 RNA is required for Dux silencing, synthesis of rRNA, and exit from the 2-cell stage. The results reveal an essential partnership between LINE1 RNA, Nucleolin, Kap1, and peri-nucleolar chromatin in the regulation of transcription, developmental potency, and ESC self-renewal” (Percharde, Lin, Yin et al. 2018, doi:10.1016/j.cell.2018.05.043).
-
Promoter-associated RNAs
Promoter-associated RNAs are mostly small (with a large class of long
noncoding RNAs possibly included among them) and originate, as the name implies, from the
promoter regions of genes.
- A promoter-associated RNA derived from an rDNA promoter has been shown to interact with a chromatin remodeling complex and also with a regulatory binding site for rRNA genes (rDNA). It apparently forms a triple-stranded structure with the DNA, which in turn is recognized by a DNA methylating enzyme, resulting in methylation of the rRNA genes and transcriptional silencing. The authors of this study suspect “a direct and possibly widespread role of RNA:DNA structures in epigenetic regulation” (Schmitz, Mayer, Postepska and Grummt 2010).
- “Transcription factors (TFs) bind specific sequences in promoter-proximal and -distal DNA elements to regulate gene transcription. RNA is transcribed from both of these DNA elements, and some DNA binding TFs bind RNA ... We show that the ubiquitously expressed TF Yin-Yang 1 (YY1) binds to both gene regulatory elements and their associated RNA species across the entire genome. Reduced transcription of regulatory elements diminishes YY1 occupancy, whereas artificial tethering of RNA enhances YY1 occupancy at these elements. We propose that RNA makes a modest but important contribution to the maintenance of certain TFs at gene regulatory elements and suggest that transcription of regulatory elements produces a positive-feedback loop that contributes to the stability of gene expression programs” (Sigova, Abraham, Ji et al. 2015, doi:10.1126/science.aad3346).
-
Transcription initiation RNAs (tiRNAs)
“Transcription initiation RNAs (tiRNAs) are nuclear localized 18
nucleotide RNAs derived from sequences immediately downstream of RNA
polymerase II transcription start sites...tiRNAs are intimately correlated
with gene expression, RNA polymerase II binding and behaviors, and
epigenetic marks associated with transcription initiation, but not
elongation” (Taft, Hawkins, Mattick and Morris 2011).
- tiRNAs are commonly found at genomic CTCF binding sites (see “Insulator protein CTCF” below) — especially when RNA polymerase II colocalizes with these sites. Evidence suggests that tiRNA helps regulate gene expression by modulating “local epigenetic structure, which in turn regulates CTCF localization”. Also, “tiRNA-regulated CTCF binding influences the levels of trimethylated H3K27 at the alternate upstream p21 promoter, and affects the levels of alternate p21 transcripts” (Taft, Hawkins, Mattick and Morris 2011).
-
tRNA-derived small RNAs (tsRNAs)
“Transfer RNA (tRNA)-derived small RNAs (tsRNAs) are among the most ancient
small RNAs in all domains of life and are generated by the cleavage of
tRNAs. Emerging studies have begun to reveal the versatile roles of tsRNAs
in fundamental biological processes, including gene silencing, ribosome
biogenesis, retrotransposition, and epigenetic inheritance, which are rooted
in tsRNA sequence conservation, RNA modifications, and protein-binding
abilities”
(Chen, Zhang, Shi et al. 2021, doi:10.1016/j.tibs.2021.05.001).
“As one of the most abundant and conserved RNA species, transfer RNAs
(tRNAs) are well known for their role in reading the codons on messenger
RNAs and translating them into proteins. In this review, we discuss the
noncanonical functions of tRNAs. These include tRNAs as precursors to novel
small RNA molecules derived from tRNAs, also called tRNA-derived fragments,
that are abundant across species and have diverse functions in different
biological processes, including regulating protein translation,
Argonaute-dependent gene silencing, and more. Furthermore, the role of tRNAs
in biosynthesis and other regulatory pathways, including nutrient sensing,
splicing, transcription, retroelement regulation, immune response, and
apoptosis, is reviewed. Genome organization and sequence variation of tRNA
genes are also discussed in light of their noncanonical functions”
(Su, Wilson, Kumar and Dutta 2020, doi:/10.1146/annurev-genet-022620-101840).
“Fragments of mature tRNAs have long been considered as mere degradation
products without physiological function. However, recent reports show that
tRNA-derived small RNAs (tsRNAs) play prominent roles in diverse cellular
processes across a wide spectrum of species. Contrasting the situation in
other small RNA pathways the mechanisms behind these effects appear more
diverse, more complex, and are generally less well understood. Here, we
provide an initial overview of tsRNA expression in different species and
tissues, revealing very high levels of 5′ tRNA halves (5′ tRHs) particularly
in the primate hippocampus. [We found evidence] suggesting that 5′ tRHs
silence genes in a sequence-specific manner, while the most efficient target
sites align to the mid-region of the 5′ tRH and are located within the CDS
[coding sequences] or 3′ UTR of the target. This amends previous
observations that tsRNAs guide Argonaute proteins to silence their targets
via a miRNA-like 5′ seed match and suggests a yet unknown mechanism of
regulation. Finally, our data suggest that some 5′ tRHs that are also able
to sequence-specifically stabilize mRNAs as up-regulated mRNAs are also
significantly enriched for 5′ tRH target sites”
(Jehn, Treml, Wulsch et al. 2020; doi:10.1261/rna.073395.119).
- “Transfer-RNA-derived small RNAs (tsRNAs; also called tRNA-derived fragments) are an abundant class of small non-coding RNAs whose biological roles are not well understood. Here we show that inhibition of a specific tsRNA, LeuCAG3' tsRNA, induces apoptosis in rapidly dividing cells in vitro and in a patient-derived orthotopic hepatocellular carcinoma model in mice. This tsRNA binds at least two ribosomal protein mRNAs (RPS28 and RPS15) to enhance their translation. A decrease in translation of RPS28 mRNA blocks pre-18S ribosomal RNA processing, resulting in a reduction in the number of 40S ribosomal subunits. These data establish a post-transcriptional mechanism that can fine-tune gene expression during different physiological states” (Kim, Fuchs, Wang et al. 2017a, doi:10.1038/nature25005).
- “tRNA related RNA fragments (tRFs), also known as tRNA-derived RNAs (tdRNAs), are abundant small RNAs reported to be associated with Argonaute proteins, yet their function is unclear. We show that endogenous 18 nucleotide tRFs derived from the 3′ ends of tRNAs (tRF-3) post-transcriptionally repress genes in HEK293T cells in culture. tRF-3 levels increase upon parental tRNA overexpression. This represses target genes with a sequence complementary to the tRF-3 in the 3′ UTR. The tRF-3-mediated repression is Dicer-independent, Argonaute-dependent, and the targets are recognized by sequence complementarity. Furthermore, tRF-3:target mRNA pairs in the RNA induced silencing complex associate with GW182 proteins, known to repress translation and promote the degradation of target mRNAs. RNA-seq demonstrates that endogenous target genes are specifically decreased upon tRF-3 induction. Therefore, Dicer-independent tRF-3s, generated upon tRNA overexpression, repress genes post-transcriptionally through an Argonaute-GW182 containing RISC via sequence matches with target mRNAs” (Kuscu, Kumar, Kiran et al. 2018, doi:10.1261/rna.066126.118).
- Recent studies “have promoted the idea that various tsRNAs produced by tRNA fragmentation can engender acrobatic ways to regulate multiple aspects of translation machinery. In particular, the function of tsRNAs is augmented by unexpected roles of RNA modifications and RNA secondary structure. Similarly, recent studies found that mammalian Nsun2- and Dnmt2-mediated m5C in tRNAs can profoundly affect tsRNA biogenesis and the structure of the resulting tsRNAs, suggesting tsRNA-mediated translational control in stem cell function, embryo development, and intergenerational epigenetic inheritance of specific acquired phenotypes. Recent systematic analyses of tRNA-modifying enzymes in budding yeast also revealed the widespread impact of noncoding RNA modifications in translational regulation and gene expression” (Shi, Zhang, Zhou and Chen 2019, doi:10.1016/j.tibs.2018.09.007).
- “Small RNAs derived from mature tRNAs, referred to as tRNA fragments or ‘tRFs,’ are an emerging class of regulatory RNAs with poorly understood functions. We recently identified a role for one specific tRF—5′ tRF-Gly-GCC, or tRF-GG—as a repressor of genes associated with the endogenous retroelement MERVL, but the mechanistic basis for this regulation was unknown. Here, we show that tRF-GG plays a role in production of a wide variety of noncoding RNAs—snoRNAs, scaRNAs, and snRNAs—that are dependent on Cajal bodies for stability and activity. Among these noncoding RNAs, regulation of the U7 snRNA by tRF-GG modulates heterochromatin-mediated transcriptional repression of MERVL elements by supporting an adequate supply of histone proteins. Importantly, the effects of inhibiting tRF-GG on histone mRNA levels, on activity of a histone 3′ UTR reporter, and ultimately on MERVL regulation could all be suppressed by manipulating U7 RNA levels. We additionally show that the related RNA-binding proteins hnRNPF and hnRNPH bind directly to tRF-GG, and are required for Cajal body biogenesis, positioning these proteins as strong candidates for effectors of tRF-GG function in vivo. Together, our data reveal a conserved mechanism for 5′ tRNA fragment control of noncoding RNA biogenesis and, consequently, global chromatin organization” (Boskovic, Bing, Kaymak and Rando 2020, doi:10.1101/gad.332783.119).
-
Enhancer RNAs
“Our study revealed that several thousand enhancers can recruit RNA
polymerase II and transcribe noncoding RNAs upon neuronal activation. The
transcripts ... have since been independently confirmed in many different
cell types and species, suggesting that eRNA synthesis is not unique to
neurons, but more likely a universal cellular mechanism involved in
governing enhancer function” (Kim, Hemberg and Gray 2015,
doi:10.1101/cshperspect.a018622).
“Whereas long noncoding RNAs undergo maturation processes such as splicing
and polyadenylation, eRNAs are shorter (>2 kb), with little evidence of
being consistently spliced or polyadenylated” (Kim, Hemberg and Gray 2015,
doi:10.1101/cshperspect.a018622).
“The explosion of high-throughput sequencing data has revealed the
complexity and diversity of the transcriptome. These data have also
unexpectedly revealed that only 1–2% of the transcriptome provides
instructions for the synthesis of functional proteins, while the remaining
98–99% gives rise to a plethora of ncRNAs [noncoding RNAs], including
transfer RNAs (tRNAs), ribosomal RNAs (rRNAs), intronic RNAs, small nuclear
(sn)RNAs, small nucleolar (sno)RNAs, microRNAs (miRNAs) and long noncoding
RNAs (lncRNAs). A recent addition to the expanding list of regulatory ncRNAs
is the emerging class of enhancer RNAs (eRNAs), which are transcribed from
enhancers in a tissue-specific manner. Increasing evidence that ncRNAs
regulate gene expression has fundamentally altered how the scientific
community views RNA-mediated gene regulation. These new advances in our
understanding of ncRNAs have also piqued an interest in pursuing
investigations of them, as the functions of the vast majority of ncRNAs
remain to be determined.”
The researchers go on to report these broad classes of enhancer RNA function:
- “eRNAs contribute to gene control by altering the chromatin environment”.
- “eRNAs interact with transcriptional regulators to control gene expression”.
- “eRNAs in tumor-promoting gene regulation and genomic instability in cancer”.
- A certain estrogen hormone (17β-oestradiol) upregulates a set of genes, and in the process causes a global increase in transcription of eRNAs from enhancers proximal to those genes. The eRNAs play a role in enhancer-promoter looping, which is stabilized by cohesin. (See “Cohesin” under THREE-DIMENSIONAL ORGANIZATION OF CHROMOSOMES, NUCLEUS, AND CELL below.) “Our data indicate that eRNAs are likely to have important functions in many regulated programs of gene transcription” (Li, Notani, Ma et al. 2013).
- “Emerging studies, showing that eRNAs function in controlling mRNA transcription, challenge the idea that enhancers are merely sites of transcription factor assembly. Instead, communication between promoters and enhancers can be bidirectional with promoters required to activate enhancer transcription. Reciprocally, eRNAs may then facilitate enhancer–promoter interaction or activate promoter-driven transcription” (Kim, Hemberg and Gray 2015, doi:10.1101/cshperspect.a018622).
- “The discovery and emerging functional roles of eRNAs certainly expand the growing regulatory capacity of noncoding RNAs. These findings not only illustrate a more complex role of cis-regulatory sequences than previously appreciated, but also provide an exciting avenue of future research in unraveling the intricate layers of gene regulation that are intertwined with lncRNAs, cis-regulatory sequences, epigenetic modifications, and three-dimensional chromatin configuration” (Kim, Hemberg and Gray 2015, doi:10.1101/cshperspect.a018622).
- Enhancer RNAs and the integrator complex: Integrator is a multi-subunit complex associated with RNA polymerase II and known to be required for 3'-end processing of certain non-polyadenylated, small nuclear RNA transcripts. It is now found to be essential for the “biogenesis of transcripts derived from distal regulatory elements (enhancers) involved in tissue- and temporal-specific regulation of gene expression in metazoans. Integrator is recruited to enhancers and super-enhancers in a stimulus-dependent manner. Functional depletion of Integrator subunits diminishes the signal-dependent induction of enhancer RNAs (eRNAs) and abrogates stimulus-induced enhancer–promoter chromatin looping ... [there is] a role for Integrator in 3′-end cleavage of eRNA primary transcripts leading to transcriptional termination. In the absence of Integrator, eRNAs remain bound to RNAPII and their primary transcripts accumulate. Notably, the induction of eRNAs and gene expression responsiveness requires the catalytic activity of Integrator complex” (Lai, Gardini, Zhang and Shiekhattar 2015, doi:10.1038/nature14906).
-
“A model is emerging in which transcription is itself an early step in
enhancer activation. Pol II is recruited by transcription factors and
maintains opens chromatin. Once the enzyme begins to transcribe, the
nascent eRNA it produces stimulates co-activator proteins such as CBP in
the region in a sequence- and stability-independent manner. The
activities of these proteins promote the recruitment of more
transcription factors, Pol II and chromatin-remodelling proteins,
enabling full enhancer activation. In addition, Pol II itself can serve
as a vehicle for attracting chromatin-modifying enzymes that spread more
molecular marks associated with chromatin activation across the
transcribed region. In this manner, transcription of enhancers can
generate a positive-feedback loop that stabilizes both enhancer activity
and gene-expression profiles.
“Overall, the current study fundamentally changes the discourse around eRNA functions, by demonstrating that these RNAs can have major, locus-specific roles in enhancer activity that do not require a particular RNA-sequence context or abundance. Furthermore, by providing strong evidence that CBP interacts with eRNAs as they are being transcribed, this study highlights the value of investigating nascent RNAs for understanding enhancer activity” (Adelman and Egan 2017, doi:10.1038/543183a).
-
“Recently, Ruthenburg and colleagues discovered a novel class of
putative eRNAs [enhancer RNAs] that remain bound to chromatin and are
not easily solubilized, which they termed chromatin-enriched enhancer
RNAs or cheRNAs.
“cheRNAs show several molecular characteristics that are distinct from those of eRNAs. Whereas most eRNAs are bidirectionally transcribed from the prototypical enhancers, cheRNAs show a specific strand bias. Moreover, eRNAs are marked by the histone H3K4 monomethylation (H3K4me1) and H3 lysine 27 acetylation (H3K27ac), whereas cheRNAs are associated with H3K4me3. Finally, cheRNAs are longer than eRNAs (median length of ~2,000 as compared to ~350 nucleotides) ... A majority of these cheRNAs remain attached to chromatin through interactions with RNA polymerase II ... cheRNAs are expressed in a cell-type-specific manner, and ... these RNAs promote changes in chromatin architecture and thereby contribute to the expression of nearby genes. cheRNA profiling in three divergent cell lines, HEK293, K562, and H1hESCs, showed that proximity to cheRNAs was a better predictor of cis-gene expression than features such as chromatin modification signatures or the expression of eRNAs or other lncRNAs” (Gayen and Kalantry 2017, doi:10.1038/nsmb.3430).
- See also Promoter-associated RNAs above.
-
Antisense RNAs
This actually belongs under almost all the main headings above. Antisense
RNA — that is, RNA transcribed from the double helix strand opposite to the
one primarily used for transcribing protein-coding RNAs — can, for example,
carry out both transcriptional and post-transcriptional regulation, and can
even code for proteins. The number of known antisense RNAs is rapidly
growing. They can be either small or very large (thousands of bases).
- “Transcription in antisense occurs with more than 70% of human coding and noncoding transcriptional units and produces NATs [natural antisense transcripts], which modulate the expression of the corresponding sense transcripts at the epigenetic, transcriptional, or posttranscriptional level” (Poliseno 2012a).
- Functionality of antisense RNAs is supported by their highly tissue-specific patterns of expression and by the fact that the promoters of active loci of antisense expression show activating histone modifications and are occupied by RNA Polymerase II, both of which are correlated with antisense RNA expression (Conley and Jordan 2012).
- In yeast, it’s been shown that 3' regions of genes can contain promoters for antisense transcription, and pre-initiation complexes (PICs) form on these promoters 60% as often as on the corresponding 5' promoters. And at these genes antisense transcription is 45% of the levels observed for sense transcription. There are suggestions that antisense transcription can occur even in the absence of sense transcription. “Our results suggest that antisense transcription can be regulated independently of divergent sense transcription in a PIC-dependent manner and we propose that regulated production of antisense transcripts represents a fundamental and widespread component of gene regulation” (Murray, Barros, Brown et al. 2012).
- Many antisense RNAs act by base-pairing with mRNAs. They may do so by extensive base-pairing with mRNAs transcribed from the DNA strand opposite their own, or by limited base-pairing with mRNAs transcribed elsewhere (and typically having different base sequences).
- “Antisense RNAs have been shown to repress target mRNAs encoding proteins, such as transposons and toxic proteins, that have the potential to be detrimental to the cell. They also have been shown to positively and negatively impact the expression of transcription regulators as well as a number of other metabolic and virulence proteins, many of which are regulated extensively at other levels” (Thomason and Storz 2010).
- Antisense RNAs seem to play a role in the structuring and restructuring of chromatin, with profound implications for gene expression. “RNA-mediated epigenetic modification has received an increasing amount of experimental support. Antisense transcripts can provide a scaffold for effector proteins to interact with DNA and chromatin in a locus-specific way”. “NATs [natural antisense transcripts] have emerged as powerful transducers of biological information, primarily due to their ability to bridge the interaction between proteins and DNA. The information content and structural features of these ncRNAs collectively establish a dynamic interace with other macromolecules, thus facilitating the formation and modulation of ribonucleoprotein complexes crucial for epigenetic signaling. These unique features permit NATs and other lncRNAs to function as scaffolds to regulate epigenetic mechanisms within the cell” (Magistri, Faghihi, St. Laurent III and Wahlestedt (2012).
-
Antisense RNAs can act by many different means, including these
(Thomason and Storz 2010):
- Interference with gene transcription: transcription of the antisense RNA in one direction blocks transcription from a promoter on the opposite strand.
- Attenuation of transcription: the antisense RNA base pairs with an RNA being transcribed, affecting the structure and therefore the transcription termination of the latter.
- Promotion or deterrence of RNA degradation: the antisense RNA can bind to an RNA transcript in such a way as to either create a target site for an enzyme that will cleave the transcript, or else block such a site, preventing cleavage.
- By base-pairing with RNA transcripts at sequences required for binding to a ribosome, antisense RNAs can directly block such binding, thereby preventing translation of the transcripts into protein. They can also indirectly affect translation (either positively or negatively) by altering transcript structure at a site distant from the base-pairing region.
-
5' and 3' untranslated regions
[Most of what might go in this section is scattered elsewhere. Try searching on either of these two strings: 5' 3'Also, see Alternative cleavage, polyadenylation, and deadenylation under Post-Transcriptional Decision-Making and Alternative coding sequences (transcription start and termination) under Decision-Making Relating to Translation.]
“A new function of 3' UTRs was discovered that does not alter the fate of
the mRNA but instead affects the newly made protein. It was shown that 3'
UTRs facilitate the formation of protein-protein interactions. They do so by
acting as scaffolds to recruit proteins to the site of translation, which
enables the formation of protein complexes with the nascent peptide chain.
Protein complex formation can then determine membrane protein localization
or protein function”. “Alternative 3' UTRs facilitate the formation of
alternative protein complexes, which can perform alternative protein
functions. This diversifies proteome function without a change in amino acid
sequence”
(Mayr 2016, doi:10.1016/j.tcb.2015.10.012).
- “About 15-35% of alternative 3' UTRs have significantly different half-lives, which may contribute to the transcriptome diversity of single cells” (Mayr 2016, doi:10.1016/j.tcb.2015.10.012).
- “Translation rates of mRNAs with alternative 3' UTRs can be differentially affected by signaling. Whereas one isoform generates basal protein levels, translation of the other is induced by signaling” (Mayr 2016, doi:10.1016/j.tcb.2015.10.012).
-
Other noncoding RNA roles
-
Many noncoding RNAs, derived from intronic and intergenic regions of
chromosomes, have been found to associate with chromatin and to
regulate the expression of neighboring genes, apparently by
participating in chromatin remodeling. How this regulation is achieved
is not yet understood (Mondal, Rasmussen, Pandey et al. 2010).
- However, it’s been found that transcription of noncoding RNAs, especially in the vicinity of gene promoters, can result in the RNAs recruiting Polycomb repressive complexes to the chromatin regions around the promoters — or elsewhere in the genome — resulting in gene repression. This is important, for example, in the silencing of cell lineage-specific genes during early development (Guenther and Young 2010).
- By forming DNA-DNA-RNA triple helixes, noncoding RNAs can inhibit promoter activity, thereby influencing expression of the associated genes.
- A new class of small RNAs — aluRNAs — has been found to play a role in formation of nucleoli, regions of the cell nucleus where chromosome loci involved in the production of ribosomal RNAs are gathered together for efficient transcription. “Splicing of pre-mRNAs containing intronic Alu elements generates short aluRNAs that associate with nucleolar proteins nucleolin and nucleophosmin. These aluRNAs, which are capable of attracting genomic loci to the nucleolus, create a scaffold that may contribute to clustering of nucleolar organizing regions (NORs) from different chromosomes via interactions with [the transcription factor] UBF.” (Carmo-Fonseca 2015, doi:10.15252/embj.201593185). Alu elements — more than a million of them in the human genome — constitute about 10% of our entire genome. They are transposons, and this finding is merely one of a continuing series of revelations about the functionality of this particular sort of “junk” DNA.
- “Hansen et al. describe a new class of short regulatory RNAs, which associate with Argonaute (AGO) proteins and derive from short introns, hence are termed agotrons. The authors annotated 87 agotrons in human and 18 in mouse, and found that they are conserved across mammalian species. Agotrons are ~80–100 nucleotides long, CG-rich and potentially form strong secondary structures. Vectors encoding three different agotrons (and their flanking exons) were transfected into human cells; the agotrons were expressed but were almost undetectable without co-expression of AGO1 or AGO2, indicating that AGO proteins stabilize spliced agotrons. Similarly to microRNAs, agotrons suppressed the expression of reporter transcripts based on seed-mediated complementarity, but their biogenesis is independent of Dicer: they associate with AGO as spliced but otherwise unprocessed introns. Agotrons potentially have a limited target repertoire compared with microRNAs but are possibly less prone to off-target effects” (Zlotorynski 2016, doi:10.1038/nrm.2016.84).
- “7SK is a small nuclear RNA (snRNA) that forms ribonucleoprotein complexes (snRNPs), which are known to regulate RNA polymerase II promoter-proximal pausing ... 7SK extensively occupies transcribed genomic regions and is particularly highly enriched at super-enhancers — regulatory regions that promote high transcriptional activity. Interestingly, at super-enhancers, 7SK associated with proteins that were distinct from the ones found in the 7SK snRNP complex at promoters and specifically recruited the chromatin-remodelling BAF complex to these sites. This 7SK-mediated BAF recruitment was shown to prevent extensive transcription at super-enhancers, which often leads to convergent mRNA synthesis (occurring simultaneously at both DNA strands) and concomitant DNA damage” (Strzyz 2016, doi:10.1038/nrm.2016.33).
- Regarding a class of small nucleolar RNAs, which can chemically modify other kinds of RNA: “SNORDs [C/D box small nucleoloar RNAs, or snoRNAs] can act to regulate pre-mRNA alternative splicing, mRNA abundance, activate enzymes, and be processed into shorter ncRNAs resembling miRNAs and piRNAs. Furthermore, recent biochemical studies have shown that a given SNORD can form both methylating and non-methylating ribonucleoprotein complexes, providing an indication of the likely physical basis for such diverse new functions. Thus, SNORDs are more structurally and functionally diverse than previously thought, and their role in gene expression is under-appreciated” (Falaleeva, Welden, Duncan and Stamm 2017, doi:10.1002/bies.201600264).
- “A study now shows that the fusion oncoprotein AML1-ETO regulates leukaemogenesis by increasing the expression of small nucleolar RNAs through post-transcriptional mechanisms, resulting in increased ribosomal RNA methylation, protein translation, and promotion of leukaemic-cell self-renewal and growth” (Khalaj and Park 2017, doi:10.1038/ncb3566).
- “Telomeres, DNA-protein complexes that protect the ends of chromosomes, were initially thought to be transcriptionally inert. However, transcripts of heterogeneous lengths containing telomeric repeats were found to originate from subtelomeric regions on several chromosomes. Since their discovery, telomeric repeat-containing RNA (TERRA) transcripts have been implicated in regulation of telomerase, the enzyme that lengthens telomeres, in the formation of heterochromatin at telomeres, and in telomere stability. Although TERRA association with telomeric chromatin has long been known, the consequences of this binding and its regulation have remained opaque. [Now, researchers] reveal differential regulation of TERRA according to the cell cycle and to telomere length, uncovering an elegant feedback loop for telomere length maintenance. Also ... TERRA binds to extra-telomeric chromatin and influences the transcription of nearby genes; additionally, TERRA binds a proteome involved in diverse processes, including chromatin remodeling and transcription” (Roake and Artandi 2017, doi:10.1016/j.cell.2017.06.020).
- “In the mouse, long terminal repeat (LTR)-retrotransposons, or endogenous retroviruses (ERV), account for most novel insertions and are expressed in the absence of histone H3 lysine 9 trimethylation in preimplantation stem cells. We found abundant 18 nt tRNA-derived small RNA (tRF) in these cells and ubiquitously expressed 22 nt tRFs that include the 3' terminal CCA of mature tRNAs and target the tRNA primer binding site (PBS) essential for ERV reverse transcription. We show that the two most active ERV families, IAP and MusD/ETn, are major targets and are strongly inhibited by tRFs in retrotransposition assays. 22 nt tRFs post-transcriptionally silence coding-competent ERVs, while 18 nt tRFs specifically interfere with reverse transcription and retrotransposon mobility. The PBS offers a unique target to specifically inhibit LTR-retrotransposons, and tRF-targeting is a potentially highly conserved mechanism of small RNA–mediated transposon control” (Schorn, Gutbrod, LeBlanc and Martienssen 2017, doi:10.1016/j.cell.2017.06.013).
-
“It is now established that [the gene] Bcl11b specifies T cell fate.
Here, we show that in developing T cells, the Bcl11b enhancer
repositioned from the lamina to the nuclear interior. Our search for
factors that relocalized the Bcl11b enhancer identified a non-coding RNA
named ThymoD (thymocyte differentiation factor). ThymoD-deficient mice
displayed a block at the onset of T cell development and developed
lymphoid malignancies. We found that ThymoD transcription promoted
demethylation at CTCF bound sites and activated cohesin-dependent looping
to reposition the Bcl11b enhancer from the lamina to the nuclear interior
and to juxtapose the Bcl11b enhancer and promoter into a single-loop
domain. These large-scale changes in nuclear architecture were associated
with the deposition of activating epigenetic marks across the loop
domain, plausibly facilitating phase separation. These data indicate how,
during developmental progression and tumor suppression, non-coding
transcription orchestrates chromatin folding and compartmentalization to
direct with high precision enhancer-promoter communication”.
Article highlights: “Non-coding transcription directs loop extrusion; non-coding transcription dictates compartmentalization; non-coding transcription directs enhancer-promoter communication; non-coding transcription establishes T cell identity and blocks lymphoid malignancy” (Isoda, Moore, He et al. 2017, doi:10.1016/j.cell.2017.09.001).
-
“• Centromeres are transcribed at a low level and transcripts are
incorporated into centromeric chromatin, where they serve essential
functions.
“• Several kinetochore proteins bind centromeric transcripts, which may be necessary to stabilize or localize the proteins.
“• Loading of centromere-specific nucleosomes may be coupled to centromeric transcription.
“• Some centromeres have known promoter activity and most centromeres are enriched in non-B form DNA that may facilitate transcription or loading of centromere-specific nucleosomes.”
“• Whereas other noncoding RNAs regulate gene expression or silence transposons, cotranscriptional assembly of kinetochores is a novel function for noncoding RNAs” (Talbert and Henikoff 2018, doi:10.1016/j.tig.2018.05.001). - “Vault RNAs (vtRNA) are small non-coding RNAs transcribed by RNA polymerase III found in many eukaryotes. Although they have been linked to drug resistance, apoptosis, and viral replication, their molecular functions remain unclear. Here, we show that vault RNAs directly bind the autophagy receptor sequestosome-1/p62 in human and murine cells. Overexpression of human vtRNA1-1 inhibits ... p62-dependent autophagy. Starvation of cells reduces the steady-state and p62-bound levels of vault RNA1-1 and induces autophagy. Mechanistically, p62 mutants that fail to bind vtRNAs display increased p62 homo-oligomerization and augmented interaction with autophagic effectors. Thus, vtRNA1-1 directly regulates selective autophagy by binding p62 and interference with oligomerization, a critical step of p62 function. Our data uncover a striking example of the potential of RNA to control protein functions directly, as previously recognized for protein-protein interactions and post-translational modifications” (Horos, Büscher, Kleinendorst, et al. 2019, doi:10.1016/j.cell.2019.01.030).
-
Many noncoding RNAs, derived from intronic and intergenic regions of
chromosomes, have been found to associate with chromatin and to
regulate the expression of neighboring genes, apparently by
participating in chromatin remodeling. How this regulation is achieved
is not yet understood (Mondal, Rasmussen, Pandey et al. 2010).
-
Caveat regarding “coding” and “noncoding” RNA
The distinction, not only between long and short noncoding RNA, but also
between noncoding RNA in general and coding RNA is increasingly being
eroded. “The coding versus non-coding classification ignores the
multifunctionality of RNA transcripts: (i) A number of so called non-coding
RNAs have open reading frames (ORFs), and it is difficult to exclude that
these may be translated at a certain development stage or in a specific
tissue. Indeed, it has been reported that peptides were translated from
rather short ORFs of some ‘non-coding’ RNAs. (ii) Coding RNAs contain a
significant amount of non-coding sequence elements in their introns and
5'-untranslated and 3'-untranslated regions (UTRs) that possess regulatory
function. Interestingly, a recent report describes the separate expression
of a large number of 3'-UTRs in human and mouse cells...(iii) While the
initial primary transcript may be ‘long’, it might be processed or a
mapping analysis might reveal that the part relevant for the activity under
investigation is significantly shorter as, for example, demonstrated for
the RNA-directed DNA methylation of ribosomal RNA (rRNA) genes. (iv) A
structural function of coding RNA transcripts in maintaining an open
chromatin structure was reported” (Caudron-Herger and Rippe 2012).
“Transcription and chromatin function are regulated by proteins that bind to
DNA, nucleosomes or RNA polymerase II, with specific non-coding RNAs
(ncRNAs) functioning to modulate their recruitment or activity. Unlike
ncRNAs, nascent pre-mRNA was considered to be primarily a passive player in
these processes ... We describe recently identified interactions between
nascent pre-mRNAs and regulatory proteins, highlight commonalities between
the functions of nascent pre-mRNA and nascent ncRNA, and propose that both
types of RNA have an active role in transcription and chromatin regulation”
(Skalska, Beltran-Nebot, Ule and Jenner 2017, doi:10.1038/nrm.2017.12).
- “The most recent analysis of 3' UTRs in mouse, human and fly indicate that a large number of 3' UTRs are expressed, in cell- and subcellular-specific manner, separately from their protein-coding sequences and function in trans as non-coding RNAs” (Kloc, Foreman and Reddy 2011).
[This is a rather silly topic. Any number of molecules not otherwise discussed in this document could be highlighted as having special, and remarkable, roles in gene regulation. Oh, well.]
Some molecules play such important and diverse roles in gene regulation that
they are worth describing in a place of their own in order to make their
importance more visible. Here I offer only one class of examples: the heat
shock proteins with chaperone functions. I will focus particularly on Hsp90
(heat shock protein 90), which is mentioned here or there elsewhere in this
document. Chaperones such as Hsp90 help proteins obtain their proper folded
conformation, help to stabilize them once they achieve this conformation, and
help to assemble and disassemble multiple-protein complexes. (Hsp90, in
cooperation with other chaperones, takes part in the assembly of RNA polymerase
II). All this helps to explain the name “chaperone”.
“Surviving extreme conditions requires the instantaneous expression of
chaperones that help to overcome stressful situations. To ensure the
preferential synthesis of these heat-shock proteins, cells inhibit
transcription, pre-mRNA processing and nuclear export of non-heat-shock
transcripts, while stress-specific mRNAs are exclusively exported and
translated” (Zander, Hackmann, Bender et al. 2016, doi:10.1038/nature20572).
“Hsp90 is an essential and abundantly expressed molecular chaperone. Although
at first sight its function seems to be restricted to the folding of
transcription factors and kinases, Hsp90 and its pool of cochaperones are
involved in virtually all cellular pathways and are implicated in a wide range
of biological processes and a myriad of diseases” (Oosten-Hawle, Bolon and
LaPointe 2016, doi:10.1038/nsmb.3359).
By ensuring proper protein folding, Hsp90 (along with other chaperone
molecules) carries out much of its regulatation of gene expression at the “end
of the line” that extends from DNA transcription to mRNA pre-processing to mRNA
translation in the cytoplasm (protein production) to the sculpting of a fully
functional protein. This activity of the chaperone is brought to bear on many
proteins. It has long been known that these functions of Hsp90 are decisively
important for the living cell.
More recently, attention has shifted to the 2–3% of total cellular Hsp90 that
is located in the nucleus, where it is a key player in gene regulation.
“Almost one-third of all genes — coding and non-coding — appear to be
influenced by the chaperone at chromatin” (Sawarkar and Paro 2013). Here I
concentrate especially on the chaperone activities that take place in the
nucleus, more or less proximal to DNA transcription.
More recently, attention has shifted to the 2–3% of total cellular Hsp90 that
is located in the nucleus, where it is a key player in gene regulation.
“Almost one-third of all genes — coding and non-coding — appear to be
influenced by the chaperone at chromatin” (Sawarkar and Paro 2013). Here I
concentrate especially on the chaperone activities that take place in the
nucleus, more or less proximal to DNA transcription.
From a recent summary of the functions of Hsp70-family proteins:
(Walters and Parker 2015, doi:10.1016/j.tibs.2015.08.004)
— “By multiple mechanisms, Hsp70 family members sense perturbation of proteostasis and then modulate aspects of mRNA metabolism”.
— “Hsp70 family members promote nascent protein folding and when defective this leads to inhibition of translation elongation”.
— “Hsp70 family members can be titrated by unfolded proteins leading to the activation of stress responsive signal transduction systems that modulate the transcriptome”.
— “Hsp70 family members can modulate the protein composition of individual mRNPs, thereby affecting their function”.
— “Hsp70 proteins promote disassembly of stress granules and are important for recovery of translation after stress”.
“Heat shock protein 90 (HSP90) is a chaperone with vital roles in regulating
proteostasis, long recognized for its function in protein folding and
maturation. A view is emerging that identifies HSP90 not as one protein that is
structurally and functionally homogeneous but, rather, as a protein that is
shaped by its environment. In this Review, we discuss evidence of multiple
structural forms of HSP90 in health and disease, including homo-oligomers and
hetero-oligomers, also termed epichaperomes, and examine the impact of stress,
post-translational modifications and co-chaperones on their formation. We
describe how these variations influence context-dependent functions of HSP90 as
well as its interaction with other chaperones, co-chaperones and proteins, and
how this structural complexity of HSP90 impacts and is impacted by its
interaction with small molecule modulators”
(Chiosis, Digwal, Trepel and Neckers 2023, doi:10.1038/s41580-023-00640-9).
- Three general principles of transcriptional regulation by Hsp90: “First, the chaperone accumulates close to the transcription start sites of one-third of all protein-coding genes and several miRNA-coding genes. Second, the Hsp90-target genes have one regulatory feature in common – they all exhibit the paused state of RNA polymerase II (pol II). Third, Hsp90 inhibition releases the pause within minutes, causing robust upregulation of many of the target genes. Evidently, one of the factors required for pol II pausing, namely the Negative Elongation Factor complex, is bound and stabilized by Hsp90 at promoters” (Sawarkar and Paro 2013).
- Some examples of the diverse roles of Hsp90: (1) “Hsp90 along with p23 is involved in disassembling the nuclear receptor complexes formed at target promoters on stimulation with ligand/hormones”; (2) “Hsp90 chaperones various proteins that act as either transcriptional repressors or activators, depending on the cell type and target genes”; (3) “Hsp90 also aids in removing nucleosomes from induced genes in yeast, facilitating transcription by RNA polymerase II”. In sum, “Hsp90 does not act as a general repressor or an activator of transcription, but rather chaperones different proteins in a gene-specific way. ... It wears different hats at different promoters” (Sawarkar and Paro 2013).
- “Given that Hsp90 plays a critical role in building the RNA pol II complex in cytosol, the chaperone may structurally assist paused or elongating pol II with the splicing machinery” (Sawarkar and Paro 2013).
- “By stabilizing a repressor called BCL-6 at promoters, Hsp90 acts to keep target genes silent in a type of B-cell lymphoma. Hsp90 inhibition in these cells results in derepression of many of these targets, including the tumor suppressor p53. Hsp90 can also activate gene expression by stabilizing and activating either transcription factors ... or epigenetic regulators” (Sawarkar and Paro 2013).
- Analyses of protein interactions “strongly suggest collaboration between Hsp90 and the transcriptional apparatus”. Hsp90 affects levels of heterochromatin protein 1 (HP1), is involved in formation of heterochromatin, and seems particularly connected also to RNA processing/splicing proteins (as well as DNA replication/damage-response proteins). “The diversity of Hsp90’s clientele may allow this chaperone functionally to couple distinct processes such as replication, DNA damage response, transcription, nuclear architecture, and splicing” (Sawarkar and Paro 2013).
- Hsp90 itself undergoes regulation, and is likely “subject to the same post-translational modifications that influence pol II and chromatin factors”. Cytosolic Hsp90 is known to be regulated by post-translational modifications, and it appears that nuclear Hsp90 may be phosphorylated, methylated, and acetylated by myriad chromatin-modifying enzymes located at promoters. “In this regard, it is noteworthy that phosphorylation of the chaperone correlates with its nuclear localization and stability” (Sawarkar and Paro 2013).
- In general, the nuclear role of Hsp90 in gene expression looks to be vastly more complex than has yet been discovered, with suggestive evidence pointing to relevant interactions of a huge variety. For example, the relationship between Hsp90 and the huge diversity of epigenetic “marks” on chromatin, and also between Hsp90 and the enzymes that apply these marks (both activating and repressive marks) appears to be intimate, but detailed processes have been hard to work out — in part because most experimental methods yield data averaged over many cells rather than showing what goes on in a single cell (Sawarkar and Paro 2013).
- How cells manage the selective retention of regular transcripts and the simultaneous rapid export of heat-shock mRNAs is largely unknown. In Saccharomyces cerevisiae, the shuttling RNA adaptor proteins Npl3, Gbp2, Hrb1 and Nab2 are loaded co-transcriptionally onto growing pre-mRNAs. For nuclear export, they recruit the export-receptor heterodimer Mex67–Mtr2 (TAP–p15 in humans). Here we show that cellular stress induces the dissociation of Mex67 and its adaptor proteins from regular mRNAs to prevent general mRNA export. At the same time, heat-shock mRNAs are rapidly exported in association with Mex67, without the need for adapters. The immediate co-transcriptional loading of Mex67 onto heat-shock mRNAs involves Hsf1, a heat-shock transcription factor that binds to heat-shock-promoter elements in stress-responsive genes. An important difference between the export modes is that adaptor-protein-bound mRNAs undergo quality control, whereas stress-specific transcripts do not. In fact, regular mRNAs are converted into uncontrolled stress-responsive transcripts if expressed under the control of a heat-shock promoter, suggesting that whether an mRNA undergoes quality control is encrypted therein. Under normal conditions, Mex67 adaptor proteins are recruited for RNA surveillance, with only quality-controlled mRNAs allowed to associate with Mex67 and leave the nucleus. Thus, at the cost of error-free mRNA formation, heat-shock mRNAs are exported and translated without delay, allowing cells to survive extreme situations” (Zander, Hackmann, Bender et al. 2016, doi:10.1038/nature20572).
Highly repetitive and mobile (transposable) DNA constitutes over 50% of the
human genome. Long disregarded as nothing but viral or freeloading
(“selfish” or “parasitic”) baggage, such sequences are now known to be
“fundamental to the cooperative molecular interactions forming
nucleoprotein complexes...The fact that repeat elements serve either as
initiators or boundaries for heterochromatin domains and provide a
significant fraction of scaffolding/matrix attachments suggests that the
repetitive component of the genome plays a major architectonic role in
higher order physical structuring”. In general, “the trend is clearly
towards discovering greater specificity, pattern and significance in the
surprisingly abundant repeat fraction of genomes” (Shapiro and von
Sternberg 2005).
Transposable elements “can be viewed as important genomic symbionts”. The
current perspective on TEs is “not as passive junk sequences nested within
larger genomes, but as important players in many biological processes in both
health and disease”. Transposons “appear to have been coopted for the purposes
of gene regulation and the orchestration of a number of processes during early
embryonic development” (Gifford, Pfaff and Macfarlan 2013).
“Building upon [Barbara] McClintock’s observations, Britten and Davidson
proposed that repetitive elements, such as transposable elements (TEs), could
provide cis-regulatory regions to an array of genes scattered throughout the
genome, allowing the coordinated control of gene expression. At first met with
skepticism and considered to be ‘junk’ or ‘selfish’ pieces of DNA, TEs have now
been shown to be major components of the genome with the ability to influence
genome evolution and function. Today TEs have been shown not only to regulate
host gene expression but are often co-opted by the host to serve new cellular
functions. It is important, however, to remember that TEs possess the ability
to hop within the genome and have the potential to cause deleterious mutations.
Therefore, it is important to keep TE mobilization ‘in check’, and host cells
have developed robust defense systems to combat unwanted TE expression and
insertion. Because TEs comprise a large part of most genomes, understanding the
role of TEs and the mechanisms by which TE expression and mobilization are
controlled is of great importance to understanding genome regulation,
variation, and evolution”
(Trends in Genetics editor, Caryn Navarro, in a special issue of the
journal on “The Mobile World of Transposable Elements”,
doi:10.1016/j.tig.2017.09.006).
“Reducing damage to the host is good for survival of TEs, because TEs, unlike
viruses, propagate mainly by vertical transfer, and thus they depend on
survival of the host. An even better strategy of a TE for survival would be to
increase the host fitness. TE proliferation is facilitated by proteins encoded
by TEs. Functions of such TE-encoded proteins include control of DNA
rearrangement (transposition and integration), transcriptional activation,
and control of chromatin states. Considering that, it may not be surprising
that TE-encoded proteins and their target cis-elements have provided rich
sources for host gene control. The beneficial effects of TEs, including
defense against other parasites, may be generated by modifications of factors
mediating their selfish behavior”
(Hosaka and Kakutani 2018, doi:10.1016/j.gde.2018.02.012).
According to new research, “The association of different repeat types with
distinct gene classes goes far beyond what has previously been shown and
suggest that such relationship might be essential for gene function and
regulation. As an example, they describe how long interspersed nuclear repeat
(LINE1) transcripts are recruited together with associated genes to silent
nuclear regions”. “Years of research have shown multiple ways in which repeats
can influence genome function. [New work] now suggests that this close
relationship between repeats and non-repetitive genes might be much more
extensive ... For example, the demonstration that LINE1 transcripts can recruit
multiple interacting proteins raises the question of what other repressors and
even activators might contribute to regulation of gene expression via LINE1
elements, as well as when and how this is achieved. Although the notion that
some repeats can recruit transcriptional regulators has been well established
for elements such as endogenous retroviruses, this work now suggests that many
other elements such as simple repeats might play an equally important role in
the regulation of gene expression.
(Zuo and Rocha 2020, doi:10.1016/j.tig.2020.03.008).
-
Transposable elements (transposons)
“Despite their classical rendition as selfish, parasitic genetic elements,
transposable elements are major drivers of genome evolution and fundamental
coordinators of regulatory function. Long suspected to harbor regulatory
roles, transposable elements have recently surfaced as conspicuous actors
behind the transcriptional and epigenomic remodeling processes underlying
development and disease. These elements are a nimble bunch, using a compact
set of molecular mechanisms in their replication yet quickly diverging in
their abundance, even among closely related species.”
(Rodriguez-Terrones and Torres-Padilla 2018, doi:10.1016/j.tig.2018.06.006)
“It was not until the 2000s, as full eukaryotic genome sequences emerged,
that we discovered that the repetitive non-coding regions of our genome
harbour large numbers of promoters, enhancers, transcription factor binding
sites and regulatory RNAs that control gene expression. More recently, the
importance of repetitive DNA in both structural and regulatory processes has
emerged, but much remains to be discovered and understood”
(Gemmell 2021, doi:10.1038/s41576-021-00354-8).
“Transposable elements (TEs) account for more than 50% of the human genome
and many have been co-opted throughout evolution to provide regulatory
functions for gene expression networks. Several lines of evidence suggest
that these networks are fine-tuned by the largest family of TE controllers,
the KRAB-containing zinc finger proteins (KZFPs). One tissue permissive for
TE transcriptional activation (termed “transposcription”) is the adult human
brain ... We reveal a distinct KZFP:TE transcriptional profile defining the
late prenatal to early postnatal transition, and the spatiotemporal and cell
type–specific activation of TE-derived alternative promoters driving the
expression of neurogenesis-associated genes. Long-read sequencing confirmed
these TE-driven [protein] isoforms as significant contributors to neurogenic
transcripts. We also show experimentally that a co-opted antisense L2
element drives temporal protein relocalization away from the endoplasmic
reticulum, suggestive of novel TE dependent protein function in primate
evolution. This work highlights the widespread dynamic nature of the
spatiotemporal KZFP:TE transcriptome and its importance throughout TE
mediated genome innovation and neurotypical human brain development”
(Playfoot, Duc, Sheppard et al. 2021, doi:10.1101/gr.275133.120 ).
“Transposable elements often contain sequences capable of recruiting the
host transcription machinery, which they use to express their own products
and promote transposition. However, the regulatory sequences carried by TEs
may affect host transcription long after the TEs have lost the ability to
transpose. Recent advances in genome analysis and engineering have
facilitated systematic interrogation of the regulatory activities of TEs ...
Notably, TEs can donate enhancer and promoter sequences that influence the
expression of host genes, modify 3D chromatin architecture and give rise to
novel regulatory genes, including non-coding RNAs and transcription factors.
We argue that TE-centric studies hold the key to unlocking general
principles of transcription regulation and evolution”
(Fueyo, Judd, Feschotte and Wysocka 2022, doi:10.1038/s41580-022-00457-y).
“Transposable elements (TEs) contribute to the evolution of gene regulatory
networks and are dynamically expressed throughout human brain development
and disease. One gene regulatory mechanism influenced by TEs is the miRNA
system of post-transcriptional control. miRNA sequences frequently overlap
TE loci and this miRNA expression landscape is crucial for control of gene
expression in adult brain and different cellular contexts ... Here, we
identify a spatiotemporally dynamic TE-embedded miRNA expression landscape
between childhood and adolescent stages of human brain development. These
miRNAs sometimes arise from two apposed TEs of the same subfamily, such as
for L2 [LINE2] or MIR [mammalian-wide interspersed repeat] elements, but in
the majority of cases stem from solo TEs. They give rise to in silico
predicted high-confidence pre-miRNA hairpin structures, likely represent
functional miRNAs, and have predicted genic targets associated with
neurogenesis. TE-embedded miRNA expression is distinct in the cerebellum
when compared to other brain regions, as has previously been described for
gene and TE expression. Furthermore, we detect expression of previously
nonannotated TE-embedded miRNAs throughout human brain development,
suggestive of a previously undetected miRNA control network. Together, as
with non-TE-embedded miRNAs, TE-embedded sequences give rise to
spatiotemporally dynamic miRNA expression networks, the implications of
which for human brain development constitute extensive avenues of future
experimental research”
(Playfoot, Sheppard, Planet and Trono 2022, doi:10.1261/rna.079100.122).
“Transposable elements (TEs) are mobile DNA elements that comprise almost
50% of mammalian genomic sequence ... [They have] had an important impact on
mammalian genome evolution and on the regulation of gene expression because
TE-derived sequences can function as cis-regulatory elements such as
enhancers, promoters and silencers. Now, advances in our ability to identify
and characterize TEs have revealed that TE-derived sequences also regulate
gene expression by both maintaining and shaping 3D genome architecture.
Studies are revealing how TEs contribute raw sequence that can give rise to
the structures that shape chromatin organization, and thus gene expression,
allowing for species-specific genome innovation and evolutionary novelty”
(Lawson, Liang and Wang 2023, doi:10.1038/s41576-023-00609-6).
- It has been proposed that “mammalian TE [transposable element] sequences [also known as ‘jumping genes’] have specific nucleosome binding properties with regulatory implications for nearby genes, are involved in the phasing of nucleosomes, and recruit epigenetic modifications to function as enhancers; that epigenetic modifications at TE sequences affect the regulation of nearby genes; and that TEs serve as epigenetic boundary elements” (Huda and Jordan 2010).
- piRNAs derived from transposons play a role in the decay of maternal RNA in the early Drosophila embryo, which in turn is crucial for proper development of the insect’s head (Rouget, Papin, Boureux et al. 2010).
- “Transposed elements [elements that have been rendered incapable of transposition through mutation] support genome integrity as part of centromeres and telomeres, affect transcription, and contribute to tissue-specific gene expression” (Singer, McConnell, Marchetto et al. 2011).
- RNAs commonly silence mobile elements by fostering the development of heterochromatin, which can then spread until it reaches an insulator. So “the insertion of a mobile element at a new genomic location typically alters the nature of chromatin surrounding the target site. Such localized chromatin alteration accounts for the phenotypic effects of many mobile element mutations. ... The majority of heterochromatic regions in eukaryotic genomes ... are rich in mobile elements” (Shapiro 2011).
- “Imprinting signals [see “Imprinting” above] are often repeats near the transcription start site, and in many cases, the repeats are clearly derived from SINEs or other mobile elements” (Shapiro 2011).
- “A total of 275,185 TSSs [transcription start sites] in human cells, representing 31.4% of all TSSs, showed homology to repeats. The majority, about 214,000, corresponded specifically to transposable elements. ... Data also suggest a high degree of spatiotemporal specificity and correlation between transposon-initiated transcription and expression of proximal genes. This suggests that coregulation of repeats and neighboring loci supersedes transcriptional interference” (Burns and Boeke 2012, referring to a study by other researchers).
- “Several studies have suggested a role [for repetitive elements] in stabilizing specific 3D genomic contacts. [We] show that the folding of the human, mouse and Drosophila genomes is associated with a significant co-localization of several specific repetitive elements, notably many elements of the SINE family. These repeats tend to be the oldest ones and are enriched in transcription factor binding sites. We propose that the co-localization of these repetitive elements may explain the global conservation of genome folding observed between homologous regions of the human and mouse genome” (Cournac, Koszul and Mozziconacci 2016, doi:10.1093/nar/gkv1292).
- “Protein interaction domains in lncRNAs can be a direct consequence of TE [transposable element] insertions because these domains are already present in TEs to mediate the assembling of ribonucleoprotein complexes necessary for the TEs’ lifecycle. These insertions can thus provide domains for interactions with proteins encoded within the TE or the genome, including transcription factors and chromatin modifiers” (Aprea and Calegari 2015, doi:10.15252/embj.201592655).
- “Additionally, TEs can provide DNA or RNA interaction domains to lncRNAs. As TEs exist as multiple copies in the genome and some of these copies form part of other transcripts in complementary orientation, each TE domain is likely capable of interacting with DNA or RNA sequences derived from the same family of TE. A lncRNA with such TE could regulate a whole family of transcripts or genomic regions” (Aprea and Calegari 2015, doi:10.15252/embj.201592655).
- Examples for the foregoing item: The ANRIL lncRNA, “encoded in a locus associated with coronary disease, acts in part by interacting with PRC1 and PRC2 while binding to the promoters of its targets in trans due to the interaction of the same Alu element (primate-specific short interspersed nuclear element) present in both the ANRIL transcript and the promoters of ANRIL-regulated genes. Another example concerning Alu elements, in this case involved in RNA-RNA interaction, is implicated in Staufen 1 (STAU1)-mediated mRNA decay. LncRNAs containing Alu elements can base-pair with an Alu element in the 3' UTR of a group of mRNAs targeted for degradation. This double-stranded RNA-RNA interaction recruits the STAU1 protein and triggers STAU1-mediated decay (Aprea and Calegari 2015, doi:10.15252/embj.201592655).
-
Retrotransposons
Some retrotransposons can be copied from one place in the genome and
inserted in another place, introducing genomic variation. They are
especially active in germ cells and during early development.
“Retrotransposition appears to be active in some somatic tissues,
including early in development, in developing neurons, and in the adult
brain, leading to mosaicism whereby different cells within an individual
have different genetic sequences”. “Ongoing retrotransposition that
results from the removal of inhibitory methylation marks on LINE and SINE
promoters is a hallmark of many cancers and also typifies neurological
disorders, including schizophrenia and Rhett syndrome”
(Elbarbary, Lucas and Maquat 2016, doi:10.1126/science.aac7247).
“Recent works demonstrating retrotransposon activity during development,
cell differentiation and neurogenesis shed new light on unexpected
activities of transposable elements”
(Mita and Boeke 2016, doi:10.1016/j.gde.2016.01.001).
“The newly gained information about retroelements made possible by great
technological advances in bioinformatics and deep sequencing leaves us
with many new questions. How does genome plasticity conferred by
retrotransposons respond to different type of environmental stresses and
what are the molecular mechanisms driving this stress-induced response?
What is the impact of retroelement mobility in processes like cancer,
cellular reprogramming and aging? What is the molecular relevance of
retrotransposon activity in tissues like the brain or developing germ
cells in which retrotransposons are not completely repressed? The more
recent perspectives on the subject seem to suggest that in these
contexts, TE [transposable element] activity can no longer be considered
simply due to spurious and uncontrolled loss of regulation because of the
newly identified ‘beneficial’ roles conferred by retrotransposons that
suggest the existence of retroelement functions co-opted and ‘safely’
modulated by the host cell. Arguably, these views leave open the idea of
‘symbiotic retrotransposons’ however antithetical this may seem to a dyed
in the wool ‘selfish gene’ devotee”
(Mita and Boeke 2016, doi:10.1016/j.gde.2016.01.001).
“Retroposed protein-coding genes are commonly considered to be
nonfunctional duplicates. However, they often gain transcriptional
capability and have important roles. Amici et al. recently identified
novel functions of a retroposed gene. HAPSTR2, a retrocopy of HAPSTR1,
encodes a protein that stabilizes the HAPSTR1 protein and functionally
buffers its loss”
(Makalowska and Kubiak 2023; doi:10.1016/j.tig.2023.03.006).
-
Retrotransposons in the germline
Large topic; not covered here (yet).
-
Some types of retrotransposons
Mouse and human studies show that “LINEs and SINEs perform many
diverse roles within cells. As DNA sequences, they can regulate
gene transcription by altering chromatin structure and by
functioning as enhancers or promoters. When transcribed as part of
a larger transcript, they can create new transcript isoforms (by
influencing alternative pre-mRNA splicing or 3'-end formation),
alter mRNA localization, change mRNA stability, tune the level of
mRNA translation, or encode amino acids that diversify the
proteome. Further, the RNA transcripts of LINEs or SINEs may
themselves function to regulate gene expression. Through their
various roles, TEs influence many aspects of cellular metabolism,
including the ability to divide, migrate, differentiate, and
respond to stress”
(Elbarbary, Lucas and Maquat 2016, doi:10.1126/science.aac7247).
“Transposable elements (TEs) are sequences currently or historically
mobile, and are present across all eukaryotic genomes. A growing
interest in understanding the regulation and function of TEs has
revealed seemingly dichotomous roles for these elements in evolution,
development, and disease. On the one hand, many gene regulatory
networks owe their organization to the spread of cis‐elements and DNA
binding sites through TE mobilization during evolution. On the other
hand, the uncontrolled activity of transposons can generate mutations
and contribute to disease, including cancer, while their increased
expression may also trigger immune pathways that result in
inflammation or senescence. Interestingly, TEs have recently been
found to have novel essential functions during mammalian development.
Here ... it is proposed that [in mammals] LINE1 is a beneficial
endogenous dual regulator of gene expression and genomic diversity
during mammalian development, and that both of these functions may be
detrimental if deregulated in disease contexts”
(Percharde, Sultana and Ramalho-Santos 2020,
doi:10.1002/bies.201900232).
-
LINE (long interspersed repeat element) retrotransposons
“Nearly 20% of human DNA is composed of LINE-1 (L1)
retrotransposons. These mobile genetic elements dominate the
evolutionary history of most mammalian genomes. Humans are no
exception: L1-encoded proteins presently orchestrate all
retrotransposition (DNA ‘copy-and-paste’ via an RNA intermediate),
as now confirmed by population-scale genomic analyses. Our genome
contains ~500,000 L1 copies, the vast majority of which are
immobile, neutrally evolving molecular fossils. A small yet
important fraction contribute to the transcriptome and gene
regulation, and to global chromatin accessibility. An even smaller
fraction, perhaps fewer than 10 L1s per person, have an appreciable
capacity to mobilize”
(Faulkner 2022, doi:10.1038/s41576-022-00485-6).
- The main class of LINEs, known as LINE-1 or L1 retrotransposons, have been found frequently to “jump,” or insert themselves “probably in many (if not most) neurons, during embryonic neuronal differentiation as well as during adult neurogenesis. This leads to neuron-to-neuron variation in genomic DNA content” (Singer, McConnell, Marchetto et al. 2010). “The consequences of L1 genomic alterations in somatic cells are still under investigation, but the high level of mutagenesis within neurons suggests that each neuron is genetically unique” (Thomas, Paquola and Muotri 2012). This recent discovery is thought to have implications for transcription in the affected neurons and therefore also for plasticity of the brain
- *** From an article entitled, “Ubiquitous L1 Mosaicism in Hippocampal Neurons”: “An estimated 13.7 somatic L1 insertions occurred per hippocampal neuron and carried the sequence hallmarks of target-primed reverse transcription. Notably, hippocampal neuron L1 insertions were specifically enriched in transcribed neuronal stem cell enhancers and hippocampus genes, increasing their probability of functional relevance” (Upton, Gerhardt, Jesuadian et al. 2015, doi:10.1016/j.cell.2015.03.026).
- “Our results demonstrate that retrotransposons [LINEs as well as some other types] mobilize to protein-coding genes differentially expressed and active in the brain. Thus, somatic genome mosaicism driven by retrotransposition may reshape the genetic circuitry that underpins normal and abnormal neurobiological processes”. Insertion of retrotransposons seems to occur particularly in the portion of the brain (the hippocampus) that is a main source of adult neurogenesis (Baillie, Barnett, Upton et al. 2011). Note: there has been debate about the actual levels of retrotransposon insertion in the brain, with possible experimental artifacts being a major concern; but note the following, more recent report.
- “Long interspersed element 1 (LINE-1 or L1) retrotransposons have generated one-third of the human genome, and their ongoing mobility is a source of inter- and intraindividual genetic diversity. Although retrotransposition in metazoans has long been considered a germline phenomenon, recent experiments using cultured cells, animal models, and human tissues have revealed extensive L1 mobilization in rodent and human neurons, as well as mobile element activity in the Drosophila brain” (Richardson, Morell and Faulkner 2014, doi:10.1146/annurev-genet-120213-092412).
- RNA transcripts of LINE elements can be incorporated directly into chromatin. “[Our] results indicate that LINE retrotransposon RNA is a previously undescribed essential structural and functional component of the neocentromeric chromatin and that retrotransposable elements may serve as a critical epigenetic determinant in the chromatin remodelling events leading to neocentromere formation” (Chueh, Northrop, Brettingham-Moore et al. (2009).
- In X chromosome inactivation (XCI), “LINEs participate in creating a silent nuclear compartment into which genes become recruited”. LINES seem to have two distinct functions in relation to XCI: silent LINEs are important for creation of the silent compartment. However, a subset of LINEs is expressed during XCI, and they play a role in propagating the inactive regions of the chromosome into neighboring areas where the genes might otherwise be prone to escape inactivation (Chow, Ciaudo, Fazzari et al. 2010).
- Among various other L1 influences: “Intronic insertions can ... impact RNA polymerase processivity through host genes, which has led to the hypothesis that L1 can act as a molecular rheostat to effect subtle changes in gene expression levels and even engage in gene breaking. The L1 antisense promoter and other transcription initiation sites in the L1 3' end can generate transcripts with the potential to impact regulation of adjacent genes. A recent study has implicated L1-derived stable nuclear RNA in regulating chromatin state, suggesting an expanded impact of L1 activity on global gene expression. L1 insertions are also subject to epigenetic regulation; in some cases, e.g., in PA-1 embryonal carcinoma cells, epigenetic marks may be targeted specifically to nascent L1 insertions during TPRT [target-site-primed reverse transcription]. Epigenetic silencing of L1 insertions may impact the expression of nearby genes if chromatin modifications spread from the L1 sequence into surrounding DNA, as seen for LTR retrotransposons. L1 retrotransposition can therefore impact the host genome, epigenome, and transcriptome via numerous routes, any of which may be sufficient to subtly or grossly alter organismal phenotype. (Richardson, Morell and Faulkner 2014, doi:10.1146/annurev-genet-120213-092412; emphasis added).
- L1 retrotransposition is not something that simply “happens to” the cell. Rather, the cell actively participates in whatever happens. “Host factors almost certainly play roles in L1 retrotransposition by interacting with the L1 mRNA-encoded proteins. Recent work by Taylor et al. identified 37 proteins that interact with the L1 RNP [ribonucleoprotein] ... Ciaudo et al. demonstrated a role for both Dicer-dependent and Ago2-dependent RNAi [RNA interference] in L1 regulation in mouse embryonic stem cells. Along with epigenetic silencing, these mechanisms defend a host genome from the likely deleterious consequences of unrestrained retrotransposition” (Richardson, Morell and Faulkner 2014, doi:10.1146/annurev-genet-120213-092412).
- “Variation of chromatin accessibility between individuals has been linked to complex traits and diseases, but the cause of only a minority of this variation is known. Now Du et al. report that transposable elements (TEs) mediate variation in chromatin accessibility in the livers of mice, resulting in differential phenotypic responses to diet. [Their experiments] suggest that certain classes of TEs — in particular, young LINEs — make a major contribution to chromatin variation. This in turn may be mediated by DNA methylation and genetic polymorphisms, and can influence metabolic phenotypes in response to diet” (Waldron 2016, doi:10.1038/nrg.2016.101).
- “Historically, LINE1 has been considered primarily as a threat to genomic integrity due to its capacity for retrotransposition and its connection to human disease. The work from Percharde et al. (2018) suggests that LINE1 elements have also evolved critical functions as lncRNAs in early development. By binding Nucleolin and KAP1, LINE1 elements facilitate both the activation of rDNA genes and the suppression of many 2C [two-cell-stage] genes via silencing of [transcription factor] Dux” (Honson and Macfarlan 2018, doi:10.1016/j.devcel.2018.06.022).
- “LINE elements recruit RNA-binding proteins to mammalian introns, influencing splicing” (blurb for doi:10.1016/ it challenging for the RNA processing machinery to identify exons accurately. We find that LINE-derived sequences (LINEs) contribute to this selection by recruiting dozens of RNA-binding proteins (RBPs) to introns. This includes MATR3, which promotes binding of PTBP1 to multivalent binding sites within LINEs. Both RBPs repress splicing and 3′ end processing within and around LINEs. Notably, repressive RBPs preferentially bind to evolutionarily young LINEs, which are located far from exons. These RBPs insulate the LINEs and the surrounding intronic regions from RNA processing. Upon evolutionary divergence, changes in RNA motifs within LINEs lead to gradual loss of their insulation. Hence, older LINEs are located closer to exons, are a common source of tissue-specific exons, and increasingly bind to RBPs that enhance RNA processing. Thus, LINEs are hubs for the assembly of repressive RBPs and also contribute to the evolution of new, lineage-specific transcripts in mammals.” (Attig, Agostini, Gooding, et al. 2018, doi:10.1016/j.cell.2018.07.001)
- “How gene expression is controlled to preserve human T cell quiescence is poorly understood. Here we show that non-canonical splicing variants containing long interspersed nuclear element 1 (LINE1) enforce naive CD4+ T cell quiescence. LINE1-containing transcripts are derived from CD4+ T cell-specific genes upregulated during T cell activation. In naive CD4+ T cells, LINE1-containing transcripts are regulated by the transcription factor IRF4 and kept at chromatin by nucleolin; these transcripts act in cis, hampering levels of histone 3 (H3) lysine 36 trimethyl (H3K36me3) and stalling gene expression. T cell activation induces LINE1-containing transcript downregulation by the splicing suppressor PTBP1 and promotes expression of the corresponding protein-coding genes by the elongating factor GTF2F1 through mTORC1. Dysfunctional T cells, exhausted in vitro or tumor-infiltrating lymphocytes (TILs), accumulate LINE1-containing transcripts at chromatin. Remarkably, depletion of LINE1-containing transcripts restores TIL effector function. Our study identifies a role for LINE1 elements in maintaining T cell quiescence and suggests that an abundance of LINE1-containing transcripts is critical for T cell effector function and exhaustion” (Marasca, Sinha, Vadalà et al. 2022, doi:10.1038/s41588-021-00989-7)
-
SINE (short interspersed repeat element) retrotransposons
“Short interspersed nuclear elements (SINEs) are nonautonomous
retrotransposons that occupy approximately 13% of the human genome.
They are transcribed by RNA polymerase III and can be
retrotranscribed and inserted back into the genome with the help of
other autonomous retroelements. Because they are preferentially
located close to or within gene-rich regions, they can regulate
gene expression by various mechanisms that act at both the DNA and
the RNA levels. In this review, we summarize recent findings on the
involvement of SINEs in different types of gene regulation and
discuss the potential regulatory functions of SINEs that are in
close proximity to genes, Pol III–transcribed SINE RNAs, and
embedded SINE sequences within Pol II–transcribed genes in the
human genome. These discoveries illustrate how the human genome has
exapted some SINEs into functional regulatory elements”
(Zhang, Pratt and Weng 2021,
doi:10.1146/annurev-genom-111620-100736).
- "So far, Alu elements [derived from SINE retrotransposons] have been documented to be cis effectors of protein-coding gene expression through their influence on transcription initiation or elongation, alternative splicing, adenosine to inosine (A-to-I) editing or translation initiation” (Gong and Maquat 2011).
- Alu RNAs “bind RNA polymerase II and repress transcription of some protein-encoding genes” (Ponicsan, Kugel and Goodrich 2010). Two such RNAs in particular “are upregulated in response to a variety of cell stresses and developmental signals. After heat shock, [they] bind directly to Pol II [RNA polymerase II] and transiently repress general transcription” (Kugel and Goodrich 2012).
- “Bi-directional transcription of a [mouse] B2 SINE establishes a boundary that places the growth hormone locus in a permissive chromatin state during mouse development” (Ponicsan, Kugel and Goodrich 2010).
- “Human mRNAs containing inverted Alu elements are present in the mammalian cytoplasm. The presence of these long intramolecular dsRNA structures within 3'-UTRs decreases translational efficiency...As inverted Alus are predicted to reside in >5% of human protein-coding genes, these intramolecular dsRNA structures are important regulators of gene expression (Capshew, Dusenbury and Hundley 2012).
- “The human genome contains about 1.5 million Alu elements, which are transcribed into Alu RNAs by RNA polymerase III. Their expression is upregulated following stress and viral infection, and they associate with the SRP9/14 protein dimer in the cytoplasm forming Alu RNPs. Using cell-free translation, we have previously shown that Alu RNPs inhibit polysome formation. Here, we describe the mechanism of Alu RNP-mediated inhibition of translation initiation and demonstrate its effect on translation of cellular and viral RNAs. Both cap-dependent and IRES-mediated initiation is inhibited. Inhibition involves direct binding of SRP9/14 to 40S ribosomal subunits and requires Alu RNA as an assembly factor but its continuous association with 40S subunits is not required for inhibition. Binding of SRP9/14 to 40S prevents 48S complex formation by interfering with the recruitment of mRNA to 40S subunits. In cells, overexpression of Alu RNA decreases translation of reporter mRNAs and this effect is alleviated with a mutation that reduces its affinity for SRP9/14. Alu RNPs also inhibit the translation of cellular mRNAs resuming translation after stress and of viral mRNAs suggesting a role of Alu RNPs in adapting the translational output in response to stress and viral infection” (Ivanova, Berger, Sherrer et al. 2015, doi:10.1093/nar/gkv048).
-
From Chen and Yang 2017, doi:10.1016/j.tcb.2017.01.002:
• “Primate-specific Alus constitute 11% of the human genome, with >1 million copies, and their genomic distribution is biased toward gene-rich regions.”
• “The functions of Alus are highly associated with their sequence and structural features.”
• “Alus can regulate gene expression by serving as cis elements.”
• “Pol-III-transcribed free Alus mainly affect Pol II transcription and mRNA translation in trans.”
• “Embedded Alus within Pol-II-transcribed mRNAs can impact their host gene expression through the regulation of alternative splicing, and RNA stability and translation.
• “Nearly half of annotated Alus are located in introns; RNA pairing formed by orientation-opposite Alus across introns promotes circRNA [circular RNA] biogenesis.”
• [Many other functional roles for Alus are also detailed by the authors.]In sum: “ the most current analyses on Alu impacts on biology are mainly focused on fixed Alu insertions in germ lines. However, Alu retrotransposon might be active in somatic tissues that continues to affect gene expression and even causes diseases, such as cancer, after birth. Thus, it will be of interest to comprehensively scrutinize how Alu insertion reshapes our genome and transcriptome in different tissues and during the lifespan in a primate-specific manner. While the impacts of some Alu repeats on the human genome have been affirmatively revealed by recent studies, the influence of other less-characterized Alus and their specific underlying mechanisms are still awaiting to be investigated. For instance, even a single point mutation in the LINE/Alu overlapped sequence of a human lncRNA could lead to lethal infantile encephalopathy. Collectively, the widespread Alu elements largely increase the complexity of gene expression and the plasticity of the human genome”.
- See also this item about aluRNAs.
- “Here we show that a conserved noncoding RNA acquires a new function due to the insertion of a mobile element. We identified a noncoding RNA, termed 5S-OT, which is transcribed from 5S rDNA loci in eukaryotes including fission yeast and mammals. 5S-OT plays a cis role in regulating the transcription of 5S rRNA in mice and humans. In the anthropoidea suborder of primates, an antisense Alu element has been inserted at the 5S-OT locus. We found that in human cells, 5S-OT regulates alternative splicing of multiple genes in trans via Alu/anti-Alu pairing with target genes and by interacting with the splicing factor U2AF65”. Splicing of more than 200 exons (about 4% of all human, alternatively spliced exons) was changed upon knockdown of 5S-OT. (Hu, Wang and Shan 2016, doi:10.1038/nsmb.3302).
- Regarding a SINE: “Overlapping gene arrangements can potentially contribute to gene expression regulation. A mammalian interspersed repeat (MIR) nested in antisense orientation within the first intron of the Polr3e gene, encoding an RNA polymerase III (Pol III) subunit, is conserved in mammals and highly occupied by Pol III ... we show that the MIR affects Polr3e expression through transcriptional interference. Our study reveals a mechanism by which a Pol II[-transcribed] gene can be regulated at the transcription elongation level by transcription of an embedded antisense Pol III gene”. “Thus, the Pol III transcribed MIR can contribute to regulation of a Pol III subunit-encoding gene” (Yeganeh, Praz, Cousin and Hernandez, 2017, doi:10.1101/gad.293324.116).
- A study of transcriptionally active B2 SINE loci in mice during a gammaherpesvirus infection “revealed transcription from 28,270 SINE loci, with ∼50% of active SINE elements residing within annotated RNA Polymerase II loci. Furthermore, B2 RNA can form intermolecular RNA-RNA interactions with complementary mRNAs, leading to nuclear retention of the targeted mRNA via a mechanism involving p54nrb. These findings illuminate a pathway for the selective regulation of mRNA export during stress via retrotransposon activation” (Karijolich, Zhao, Alla and Glaunsinger 2017, doi:10.1093/nar/gkx180)
-
LTRs (long terminal repeats)
- “We have performed deep profiling of the nuclear and cytoplasmic transcriptomes of human and mouse stem cells, identifying a class of previously undetected stem cell–specific transcripts. We show that long terminal repeat (LTR)-derived transcripts contribute extensively to the complexity of the stem cell nuclear transcriptome. Some LTR-derived transcripts are associated with enhancer regions and are likely to be involved in the maintenance of pluripotency”. “This study, together with recent reports, has probably just begun to unravel the set of unexpected functions of retrotransposons in stem cell biology” (Fort, Hashimoto, Yamada et al. 2014).
-
LINE (long interspersed repeat element) retrotransposons
- Regarding the discovery of megabase-sized functional chromosome domains: “We observed that Alu/B1 and B2 SINE retrotransposons in mouse and Alu SINE elements in humans are enriched at boundary regions. In light of recent reports indicating that a SINE B2 element functions as a boundary in mice, and SINE element retrotransposition may alter CTCF binding sites during evolution, we believe that this contributes to a growing body of evidence indicating a role for SINE elements in the organization of the genome” (Dixon, Selvaraj, Yue et al. 2012).
- Parenthetically: The evolutionary expansion of CTCF-binding sites via transposable elements has played a key role in structuring the genome. See “Insulator protein CTCF (CCCTC-binding factor)” below.
-
Retrotransposons in the germline
-
Tandem repeats
“STRs [short tandem repeats] are highly multiallelic and may contribute more
de novo mutations than any other variant class. Recent studies ...
show that STRs play a widespread role in regulating gene expression and
other molecular phenotypes. These analyses suggest that STRs are an
underappreciated but rich reservoir of variation that likely make
significant contributions to Mendelian diseases, complex traits, and cancer”
(Gymrek 2017, doi:10.1016/j.gde.2017.01.012).
“Short tandem repeats (STRs) have been implicated in a variety of complex
traits in humans. However, genome-wide studies of the effects of STRs on
gene expression thus far have had limited power to detect associations and
provide insights into putative mechanisms. Here, we leverage whole-genome
sequencing and expression data for 17 tissues from the Genotype–Tissue
Expression Project to identify more than 28,000 STRs for which repeat number
is associated with expression of nearby genes (eSTRs) ... We identify
hundreds of eSTRs linked with published genome-wide association study
signals and implicate specific eSTRs in complex traits, including height,
schizophrenia, inflammatory bowel disease and intelligence”
(Fotsing, Margoliash, Wang et al. 2019, doi:10.1038/s41588-019-0521-9).
“Variable number tandem repeats (VNTRs) make up ∼3% of the human genome but
are often excluded from association analysis owing to poor read mappability
or divergent repeat content ... We find that 9422 out of 39,125 VNTRs are
associated with nearby gene expression through motif variations ....
Fine-mapping identifies 174 genes to be likely driven by variation in
certain VNTR motifs and not overall length”
(Lu, Smaruj, Fudenberg et al. 2023; doi:10.1101/gr.276768.122).
“Approximately 5% of the human genome consists of short tandem repeats
(STRs), sequences in which repeating units of 1 to 6 base pairs form an
array of up to 100 base pairs long. Variations in STR length are
associated with, and often the causative agents of, gene expression changes
present in some hereditary conditions, for example, Huntington's disease,
autism, and schizophrenia ... Horton et al. demonstrate that STRs exert
their effects by directly binding transcription factor proteins, thus
explaining how STRs might influence gene expression in both normal and
diseased states”
(Kuhlman 2023, doi:10.1126/science.adk205)
“Tandem repeats are a large source of genetic variation but are challenging
to analyse and have been missing from most genome-wide studies. Results now
suggest that systematic incorporation of tandem repeats into complex trait
analyses is likely to yield a rich source of causal variants and new
biological insights”
(Lamkin and Gymrek 2024, doi:10.1038/s41576-024-00736-8).
- “We propose that TNRs [trinucleotide or microsatellite repeats] have potential to be functional genetic elements and that their variation may be involved in the regulation of many common phenotypes”. Current researches “argue against the common notion that microsatellites are ‘genetic junk’” (Kozlowski, de Mezer and Krzyzosiak 2010).
- “We...know that [trinucleotide] repeats having hairpin structure forming potential are over-represented in exons and therefore are likely implicated in some specific biological functions. At present, however, the normal functions of TNRs in transcripts are very poorly understood” (Krzyzosiak, Sobczak, Wojciechowska et al. 2012).
- In yeast, “as many as 25% of all gene promoters contain tandem repeat sequences ... Variations in repeat length result in changes in expression and local nucleosome positioning. Tandem repeats are variable elements in promoters that may facilitate evolutionary tuning of gene expression by affecting local chromatin structure” (Vinces, Legendre, Caldara et al. 2009).
- “Long triplet repeat RNA [acts] as a pathogenic agent by presenting human neurological diseases caused by triplet repeat expansions in which mutant RNA gains a toxic function. Prominent examples of these diseases include myotonic dystrophy type 1 and fragile X-associated tremor ataxia syndrome”. Also, there appears to be “RNA-mediated pathogenesis in polyglutamine disorders such as Huntington’s disease and spinocerebellar ataxia type 3, in which expanded CAG repeats may act as an auxiliary toxic agent” (Krzyzosiak, Sobczak, Wojciechowska et al. 2012).
- It has been shown in fruit flies that the highly variable satellite repeats on the Y chromosome can affect gene expression, apparently by altering the balance between open and condensed chromatin. Satellite repeats are binding sites for many chromatin-regulating proteins. Moreover, variations in these Y chromosome repeats seem to affect expression of thousands genes on other chromosomes, via processes that are not understood. The affected genes include many that are involved in transcription, chromatin assembly, and chromosome organization, among other things (Lemos, Branco and Hartl 2010).
- “One potential source of ‘missing heritability’ lies in variants such as STRs [short tandem repeats] that are not accessible from traditional SNP [single nucleotide polymorphism] arrays, as has been hypothesized by a growing number of groups. Concrete examples of this phenomenon are now being discovered. For instance, systematic dissection of the strongest schizophrenia association signal revealed a recurrent copy number variation not in strong LD [linkage disequilibrium] with any single SNP to be the causal variant” (Gymrek 2017, doi:10.1016/j.gde.2017.01.012).
- “While most well-studied STRs lie in or near protein coding regions, it is becoming increasingly clear that STRs located in non-coding regions play an important regulatory role. Dozens of single gene studies have identified STRs that control expression of nearby genes via a variety of mechanisms ... Furthermore, genome-wide analyses revealed that STRs are enriched in human promoter and enhancer regions and are a hallmark of enhancers in Drosophila ... STRs were shown to contribute a median of 10-15% of cis heritability of gene expression mediated by all common variants in lymphoblastoid cell lines. Taken together, these studies point to an important regulatory role of STRs, strongly supporting the hypothesis that they will contribute to complex traits in humans” (Gymrek 2017, doi:10.1016/j.gde.2017.01.012).
We see countless images of the iconic double helix, but while it does comprise
the majority of chromosomal DNA in cells, “alternative conformations (including
left-handed DNA, three-stranded triplex DNA, four-armed cruciforms,
slipped-strand DNA with two three-armed junctions, four-stranded G-quadruplex
structures and stable, unpaired helical regions) can exist in the context of
chromosomes. Rather than being a static helix, DNA possesses dynamic
flexibility and variability, as evidenced by helix regions that can be curved,
straight or flexible. Differences result from variations in base stacking and
twist angles inherent in different DNA sequences. DNA supercoiling,
particularly unrestrained supercoiling, plays a major part in the dynamic
flexibility and topological contortions of the DNA double helix”. All this
bears directly on gene expression — for example, by “tuning the helix to
optimize sequence-specific recognition by a protein” (Sinden 2013).
“The picture emerging is of a genome being a complex regulatory landscape. ...
We argue [that] transcriptional control over distance can be understood when
considering action in the context of the folded genome. Genome topology is
expected to differ between individual cells, and this may cause variegated
expression” (Splinter and de Laat 2011). “What was previously known as junk
DNA in fact appears a regulatory jungle. In order to understand the laws of
the jungle, linear information must now be converted into spatial
relationships. For this, highly detailed 3D topology maps need to be generated
for all regulatory sites individually” (Splinter and de Laat 2011).
“The emerging picture is one of extensive self-enforcing feedback between
activity and spatial organization of the genome, suggestive of a
self-organizing and self-perpetuating system that uses epigenetic dynamics to
regulate genome function in response to regulatory cues and to propagate
cell-fate memory” (Cavalli and Misteli 2013).
“Chromatin folding and establishment of 3D genome architecture is thought to
occur downstream of the initial targeting of TFs [transcription factors] and
chromatin-modifying complexes. A recent study challenges this dogma and
suggests that the 3D genome architecture of Polycomb-associated [DNA]
topological domains can influence the binding of specific chromatin factors to
the DNA: a comparative genomics study in Drosophila species demonstrated that
sequence-specific binding of the sequence-specific DNA-binding protein PHO
outside a Polycomb context requires the presence of strong Pho consensus
motifs. By contrast, within Polycomb domains PHO is able to bind to genomic
sites containing far weaker motifs. Notably, these sites participate in
frequent chromatin interactions, consistent with known looped interactions
between PREs [Polycomb response elements in DNA]. By contrast, similar genomic
regions outside Polycomb domains show much lower contact frequencies and no Pho
binding. This suggests that the 3D association of genomic sites within Polycomb
domains stabilizes the binding of a TF. Therefore, [larger-scale] nuclear
architecture [of the sort facilitated by Polycomb group proteins] can have a
regulatory function in TF binding, similar to local chromatin structure (such
as nucleosome positioning or chromatin compaction). Future work will show
whether this finding reflects a specific feature of Polycomb domains or whether
it might apply to other chromatin factors and topologically associating
domains” (Entrevan, Schuettengruber and Cavalli 2016,
doi:10.1016/j.tcb.2016.04.009).
Key points regarding X chromosome inactivation: “Spatial interactions
between RNA, architectural factors and chromatin have essential roles during
X-chromosome inactivation; CTCF is a versatile factor that regulates chromosome
counting, allelic pairing, allelic choice and X-chromosome architecture; Xist
RNA determines the 3D structure of the inactive X chromosome by evicting
architectural proteins; the active X chromosome is organized into more than
100 topologically associated domains (TADs), whereas the inactive X chromosome
is partitioned into two megadomains; spatial partitioning of the
X-inactivation centre into two TADs allows proper Xist and Tsix expression
during X-chromosome inactivation; perinucleolar localization of the inactive X
chromosome helps to maintain its epigenetic state” (Jégu, Aeby and Lee 2017,
doi:10.1038/nrg.2017.170). See also X chromosome
inactivation above.)
“Chromosomal architecture is known to influence gene expression, yet its role
in controlling cell fate remains poorly understood. Reprogramming of somatic
cells into pluripotent stem cells (PSCs) by the transcription factors (TFs)
OCT4, SOX2, KLF4 and MYC offers an opportunity to address this question ...
Here, we ... integrate time-resolved changes in genome topology with gene
expression, TF binding and chromatin-state dynamics. The results showed that
TFs drive topological genome reorganization at multiple architectural levels,
often before changes in gene expression. Removal of locus-specific topological
barriers can explain why pluripotency genes are activated sequentially, instead
of simultaneously, during reprogramming. Together, our results implicate genome
topology as an instructive force for implementing transcriptional programs and
cell fate in mammals”
(Stadhouders, Vidal, Serra et al. 2018, doi:10.1038/s41588-017-0030-7).
“In breast cancer cells, some topologically associating domains (TADs) behave
as hormonal gene regulation units, within which gene transcription is
coordinately regulated in response to steroid hormones. Here we further
describe that responsive TADs contain 20- to 100-kb-long clusters of
intermingled estrogen receptor (ESR1) and progesterone receptor (PGR) binding
sites, hereafter called hormone-control regions (HCRs). In T47D cells, we
identified more than 200 HCRs, which are frequently bound by unliganded ESR1
and PGR. These HCRs establish steady long-distance inter-TAD interactions
between them and organize characteristic looping structures with promoters in
their TADs even in the absence of hormones in ESR1+-PGR+ cells. This
organization is dependent on the expression of the receptors and is further
dynamically modulated in response to steroid hormones. HCRs function as
platforms that integrate different signals, resulting in some cases in opposite
transcriptional responses to estrogens or progestins. Altogether, these results
suggest that steroid hormone receptors act not only as hormone-regulated
sequence-specific transcription factors but also as local and global genome
organizers”
(Dily, Vidal, Cuartero et al. 2018, https://doi.org/10.1101/gr.243824.118).
“We report cell-type specialized 3D chromatin structures at multiple genomic
scales that relate to patterns of gene expression. We discover extensive
‘melting’ of long genes when they are highly expressed and/or have high
chromatin accessibility. The contacts most specific of neuron subtypes contain
genes associated with specialized processes, such as addiction and synaptic
plasticity, which harbour putative binding sites for neuronal transcription
factors within accessible chromatin regions. Moreover, sensory receptor genes
are preferentially found in heterochromatic compartments in brain cells, which
establish strong contacts across tens of megabases. Our results demonstrate
that highly specific chromatin conformations in brain cells are tightly related
to gene regulation mechanisms and specialized functions”
(Winick-Ng, Kukalev, Harabula et al. 2021, doi:10.1038/s41586-021-04081-2).
“Liu and colleagues surveyed the epigenomic landscape across the hierarchy of
chromatin organizations during human stem cell aging, revealing how
multifaceted genomic conformational changes associated with aging converge to
impact upon senescence, and identified the activation of placenta-specific
genes as a hallmark of human cellular aging” (table of contents blurb for
Liu, Ji, Ren et al. 2022, doi:10.1016/j.devcel.2022.05.004).
(Many topics covered elsewhere in this document bear on the role of three-dimensional structure in gene regulation.)
-
Chromosome looping and long-distance chromatin interaction
Stretches of chromosomes can form loops, and sometimes these loops extend
out from their main territory in the nucleus. This looping can have
various roles, the relation between which is not clear.
Recent research identified over 1800 loops in mouse embryonic stem cells,
of which 5/6 connected loci on the same chromosome and the rest connected
different chromosomes. These “likely represent just a small fraction of
all the chromatin loops in the nucleus” (Espinoza and Ren 2011).
In chickens, “the difference in β-keratin genes expressed in feathered and
scaly skin is regulated via typical enhancers, while differential expression
within individual feathers correlates with chromatin looping within the gene
cluster” (Xu and Millar 2020, doi:10.1016/j.devcel.2020.05.008).
“Three-dimensional (3D) chromatin structure has been shown to play a role in
regulating gene transcription during biological transitions ... Changes to
epigenetic features between — rather than at — boundaries were highly
predictive of changes in looping. Together these data suggest that although
CTCF and RAD21 may be the core machinery dictating where loops form, other
features (both at the anchors and within the loop boundaries) may play a
larger role than previously anticipated in determining the relative loop
strength across cell types and conditions”
(Bond, Davis, Quiroga et al. 2023, doi:10.1101/gr.277397.122).
“Contacts between enhancers and promoters are thought to relate to their
ability to activate transcription ... We show that active regulatory
elements, independent of cohesin and polycomb, interact with each other
across distances of tens of megabases in vertebrate and invertebrate genomes
and that interactions correlate and change with activity. However, these
ultra-long-range interactions are not dependent on RNA polymerase II
transcription or individual transcription cofactors. Using simulations, we
show that a model of chromatin and multivalent binding factors can give rise
to long-range interactions via bridging-induced clustering. We propose that
long-range interactions between cis-regulatory elements are driven by at
least three distinct processes: cohesin-mediated loop extrusion, polycomb
contacts, and clustering of active regions”
(Friman, Flyamer, Marenduzzo et al. 2023, doi:10.1101/gr.277567.122).
- “Reprogramming” of differentiated cells into induced pluripotent stem cells (iPSCs) causes “major, widespread alterations to chromosome structure. The founder cell types were highly distinct from each other in cluster analyses, whereas the iPSCs were highly similar regardless of their cell of origin or passage number, and they displayed a pluripotency-associated contact signature that was also shared by embryonic stem cells ... reprogramming caused a convergence in the structure of local topologically-associated domains and near-complete dissolution of cell-type-specific chromosomal loops while increasing looping and contacts between pluripotency-associated genes” (Burgess 2016, doi:10.1038/nrg.2016.35).
- However, “despite the global convergence of chromosome structure towards a common pluripotency-associated conformation, the investigators found that iPSCs displayed subtle chromosome structure signatures in early-passage iPSCs that allowed their cell-type-of-origin to be determined. These distinguishing signatures involved intra-TAD connectivities and DNA-binding positions of the TAD-organizing protein CTCF. Intriguingly, although these signatures allowed the cell of origin to be determined, they were absent in the original source cell types. Thus, the authors [of the study being reported on] propose that these chromatin structure signatures arise de novo during reprogramming, in contrast to the ‘memory’ nature of DNA methylation and transcriptomic signatures that are a retained remnant of their cellular history” (Burgess 2016, doi:10.1038/nrg.2016.35).
- Distant enhancers may interact directly with gene promoters, thereby making a loop out of the intervening sequence. Contacts between enhancers and promoters can also happen between an enhancer and a promoter on different chromosomes.
- There is “growing evidence for physical interactions between distant loci other than enhancer-promoter juxtapositions” (Woodcock and Ghosh 2010).
- A gene promoter can make contact with the opposite (3') end of the gene, forming a loop that could encourage rapid and repeated transcription passes by RNA polymerase; when the polymerase reached the end of the gene, it would be positioned over the promoter for another round of transcription.
- In general, RNA polymerases “are often found at the cross-roads maintaining loops” and might themselves be ties helping to maintain such loops (Papantonis and Cook 2010)
- Clusters of Hox genes, which play a crucial role in development, have been found organized into loops, with the loop pattern differing for each cluster. The looping is associated with the silent state of the genes. Activation of the genes involves “extensive nuclear reorganization” (Ferraiuolo, Rousseau, Miyamoto et al. 2010). In flies, silencing of clustered Hox genes occurs in conjunction with polycomb group proteins.
- Polycomb group proteins and the complexes they form with other factors play a role in long-range contacts between chromosome loci. “Recent work has clearly shown that PcG [Polycomb group] proteins as well as other nuclear factors organize complex 3D chromosome-folding patterns that impinge on gene expression, such that we can no longer ignore chromatin architecture when studying the regulation of any genomic locus” (Bantignies and Cavalli 2011).
-
A study of local chromosome organization at both active and repressed genes in embryonic stem cells revealed that “super-enhancer-driven genes generally occur within chromosome structures that are formed by the looping of two interacting CTCF sites co-occupied by cohesin. (See figure at right.) These looped structures form insulated neighborhoods whose integrity is important for proper expression of local genes. We also find that repressed genes encoding lineage-specifying developmental regulators occur within insulated neighborhoods. These results provide insights into the relationship between transcriptional control of cell identity genes and control of local chromosome structure.” (doi:10.1016/j.cell.2014.09.030)
Image from Dowen, Jill M., Zi Peng Fan, Denes Hnisz et al. (2014). “Control of Cell Identity Genes Occurs in Insulated Neighborhoods in Mammalian Chromosomes”, Cell vol. 159, no. 2 (Oct. 9), pp. 374-87. doi:10.1016/j.cell.2014.09.030
- Sections of chromosomes may loop outward to join various other elements in “transcription factories”. See “Colocalization of genes” below.
- “We mapped long-range chromatin interactions associated with RNA polymerase II in human cells and uncovered widespread promoter-centered intragenic, extragenic [promoter to distal regulatory elements such as enhancer], and intergenic [promoter-promoter of different genes] interactions. These interactions further aggregated into higher-order clusters, wherein proximal and distal genes were engaged through promoter-promoter interactions. Most genes with promoter-promoter interactions were active and transcribed cooperatively, and some interacting promoters could influence each other implying combinatorial complexity of transcriptional controls. Comparative analyses of different cell lines showed that cell-specific chromatin interactions could provide structural frameworks for cell-specific transcription, and suggested significant enrichment of enhancer-promoter interactions for cell-specific functions” (Li, Ruan, Auerbach et al. 2012).
- Gene loops — which are not static, but form transiently in a transcription-dependent manner — play a role in regulation of noncoding RNA: they repress antisense transcription from bidirectional promoters (Hampsey 2012).
- “‘High-resolution identification of cohesin-mediated long-range chromatin interactions was critical for us to find the loops between two CTCF (CCCTC-binding factor) sites bracketing a domain that harbours a super-enhancer-driven pluripotency gene or Polycomb-repressed differentiation gene,’ reflects [Keji] Zhao. Not only did the team find that genes regulated by super-enhancers occur within large looped chromosome structures that are connected through interacting CTCF sites co-occupied by cohesin but, more importantly, they also showed that higher-order chromatin organization is essential for the proper regulation of gene expression. ‘CTCF and cohesin organize the loops in such a way that protects key cell identity genes from dysregulation by other regulatory elements outside the domain,’ explains Zhao. In other words, the 'super-enhancer domains' restrict super-enhancer activity to genes within the domain, as evidenced by the fact that loss of a boundary delineated by CTCF resulted in the inappropriate activation of genes located outside that boundary. ‘Many of the loops are retained throughout differentiation,’ comments [Richard A.] Young, ‘so in this study we define the chromosome structures that are the foundation for differentiation into the broad range of cell types found in mammals.’”
- Relation between chromosome loops and splicing: “Different types of chromatin contact behaviors and loops coexist in different cell types. Surprisingly, we find that the bodies of highly expressed genes interact strongly both in cis and in trans to form clusters of loops. These interactions are strongly correlated with the number of splicing events per gene with the strongest contacts occurring between genes that undergo most splicing. Splicing foci have been observed in live cells, but whether the contacts we observed are directly linked to co-transcriptional splicing remains to be seen” (Bonev, Cohen, Szabo et al. 2017, doi:10.1016/j.cell.2017.09.043).
- “We show that cohesin suppresses compartments but is required for TADs and loops, that CTCF defines their boundaries, and that the cohesin unloading factor WAPL and its PDS5 binding partners control the length of loops. In the absence of WAPL and PDS5 proteins, cohesin forms extended loops, presumably by passing CTCF sites, accumulates in axial chromosomal positions (vermicelli), and condenses chromosomes. Unexpectedly, PDS5 proteins are also required for boundary function. These results show that cohesin has an essential genome‐wide function in mediating long‐range chromatin interactions and support the hypothesis that cohesin creates these by loop extrusion, until it is delayed by CTCF in a manner dependent on PDS5 proteins, or until it is released from DNA by WAPL” (Wutz, Várnai, Nagasaka et al. 2017, doi:10.15252/embj.201798004).
- “The prevailing view of metazoan gene regulation is that individual genes are independently regulated by their own dedicated sets of transcriptional enhancers. Past studies have reported long-range gene–gene associations, but their functional importance in regulating transcription remains unclear. Here we used quantitative single-cell live imaging methods to provide a demonstration of co-dependent transcriptional dynamics of genes separated by large genomic distances in living Drosophila embryos. We find extensive physical and functional associations of distant paralogous genes, including co-regulation by shared enhancers and co-transcriptional initiation over distances of nearly 250 kilobases. Regulatory interconnectivity depends on promoter-proximal tethering elements, and perturbations in these elements uncouple transcription and alter the bursting dynamics of distant genes, suggesting a role of genome topology in the formation and stability of co-transcriptional hubs. Transcriptional coupling is detected throughout the fly genome and encompasses a broad spectrum of conserved developmental processes, suggesting a general strategy for long-range integration of gene activity” (Levo, Raimundo, Bing et al. 2022, doi:10.1038/s41586-022-04680-7).
- For more on chromosome looping, see Insulator protein CTCF (CCCTC-binding factor) and Cohesin below.
-
Chromosome domains
The chromosome is structured into domains in a variety of ways, and this
structuring is integral to the functioning of the chromosomes, including
gene expression.
“Our imaging data revealed TAD-like structures with globular conformation
and sharp domain boundaries in single cells. The boundaries varied from cell
to cell, occurring with nonzero probabilities at all genomic positions but
preferentially at CCCTC-binding factor (CTCF)- and cohesin-binding sites.
Notably, cohesin depletion, which abolished TADs at the population-average
level, did not diminish TAD-like structures in single cells but eliminated
preferential domain boundary positions. Moreover, we observed widespread,
cooperative, multiway chromatin interactions, which remained after cohesin
depletion”
(Bintu, Mateo, Su et al. 2018, doi:10.1126/science.aau1783).
“Studies of 3D chromatin organization have suggested that chromosomes are
hierarchically organized into large compartments composed of smaller domains
called topologically associating domains (TADs). Recent evidence suggests
that compartments are smaller than previously thought and that the
transcriptional or chromatin state is responsible for interactions leading
to the formation of small compartmental domains in all organisms. In
vertebrates, CTCF forms loop domains, probably via an extrusion process
involving cohesin. CTCF loops cooperate with compartmental domains to
establish the 3D organization of the genome. The continuous extrusion of the
chromatin fibre by cohesin may also be responsible for the establishment of
enhancer–promoter interactions and stochastic aspects of the transcription
process. These observations suggest that the 3D organization of the genome
is an emergent property of chromatin and its components, and thus may not be
only a determinant but also a consequence of its function”
(doi:10.1038/s41576-018-0060-8).
“[The authors of a study] find that TAD-like chromatin domain structures are
present in single cells. Although the boundaries of these TAD-like
structures differ between cells and could be present at all locations in the
genome, they do appear to be preferentially placed near CTCF and cohesin
binding sites. However, TAD-like structures are still present in single
cells even when cohesin is depleted, an observation that is not evident from
existing studies of chromatin at the population-average level. In having
this more expansive and intricate view of chromatin organization, the
authors reveal that three-way interactions between chromatin segments are
commonplace” (Kruger 2018, doi:10.1016/j.cell.2018.11.001).
“We report long-range TAD–TAD interactions that form constitutive and
variable TAD cliques. A differentiation-coupled relationship between TAD
cliques and lamina-associated domains suggests that TAD cliques stabilize
heterochromatin at the nuclear periphery. We also provide evidence of
dynamic TAD cliques during mouse embryonic stem-cell differentiation and
somatic cell reprogramming and of inter-TAD associations in single-cell
high-resolution chromosome conformation capture (Hi-C) data. TAD cliques
represent a level of four-dimensional genome conformation that reinforces
the silencing of repressed developmental genes”
(Paulsen, Ali, Nekrasov, et al. 2019, doi:10.1038/s41588-019-0392-0).
“The best-understood domain borders in mammals are those established by the
transcription factor CTCF, which blocks loop extrusion by cohesin proteins
Deleting CTCF sites at such boundaries typically results in chromosome
contacts spreading across the deleted sites, thus indicating that these
boundaries are actively defined. Acute CTCF depletion abrogates insulation
at most boundaries, thus homogenizing chromosome folding. However, not all
CTCF sites create equally sharp boundaries, and approximately 20% of borders
do not depend on CTCF, thereby indicating that other factors are also
involved” (Anderson and Nora 2020, doi:10.1038/s41588-020-0704-4).
- “The entire genome [that is, each chromosome] is linearly partitioned into well-demarcated physical domains that overlap [correlate] extensively with active and repressive epigenetic marks. Chromosomal contacts are hierarchically organized between domains ... inactive domains are condensed and confined to their chromosomal territories, whereas active domains reach out of the territory to form remote intra- and interchromosomal contacts” (Sexton, Yaffe Kenigsberg et al. 2012).
- Regarding one particular scale of organization: “Mammalian chromosomes are segmented into megabase-sized topological domains ... Such spatial organization seems to be a general property of the genome: it is pervasive throughout the genome, stable across different cell types and highly conserved between mice and humans. ... We have identified multiple factors that are associated with the boundary regions separating topological domains, including the insulator binding factor CTCF, housekeeping genes and SINE elements” (Dixon, Selvaraj, Yue et al. 2012). There is a “striking correlation between domain boundaries and cohesin/CTCF binding” (Feig and Odom 2013, reporting on work by Sofueva, Yaffe, Chan et al. 2013).
- Another group of researchers looked at the X chromosome in mice and discovered “a series of discrete 200-kilobase to 1Mb topologically associating domains (TADs), present both before and after cell differentiation and on the active and inactive X. TADs align with, but do not rely on, several domain-wide features of the epigenome, such as H3K27me3 or H3K9me2 blocks and lamina-associated domains. TADs also align with coordinately regulated gene clusters. Disruption of a TAD boundary causes ectopic chromosomal contacts and long-range transcriptional misregulation” (Nora, Lajoie, Schulz et al. 2012). The authors showed that “TAD boundaries can have a critical role in high-order chromatin folding”.
- Topological domains “cluster into transcriptionally active and inactive regions. ... Passive domains are larger and characterized by homogenous internal structures, whereas active domains are smaller yet contain more internal contact complexity”. “Active domains contain more cohesin/CTCF co-bound sites, thus suggesting an explanation for their enhanced complexity” (Feig and Odom 2013).
- “Large cell-to-cell heterogeneity in intra-TAD structure and contacts” has been found. Some loci within a TAD have an outsized role in maintaining folding structure and contacts, and these loci are enriched in CTCF and cohesin binding sites. Also, there are “correlations between the TAD compactness and the levels of nascent RNAs transcribed from [those regions], which indicated that the variable chromosome conformations have consequences for gene expression. ‘These fluctuating structures may be exploited to provide variability that can participate in setting up monoallelic gene expression (in the case of X chromosome inactivation) or differential gene expression states (in a developmental context)’, proposes [senior researcher Edith] Heard” (Burgess 2014).
- It turns out that there are functional domains of all different sizes, and different sizes have different characteristics. One research team looked at seven TADs in two mammalian cell types (embryonic stem cells and neural progenitor cells) and found over 60 sub-domains. Of 260 long-range interactions common to the two cell types, 83 were specific to embryonic stem cells and 165 were specific to neural progenitor cells. So while larger domains are typically invariant across cell types, many smaller domains are variable. The researchers also show that while chromosome looping (and associated chromatin modifications) correlate with some changes in gene expression during cell differentiation, they do not correlate with all such changes, so other factors are involved (Phillips-Cremins, Sauria, Sanyal et al. 2013; Bodnar and Spector 2013).
- The CTCF and cohesin structural proteins, as well as Mediator (a member of the pre-initiation complex — see “Pre-initiation complex” under PRE-TRANSCRIPTIONAL DECISION-MAKING above) appear to be essential for the formation of the smaller-scale chromosome domains — but in different combinations for different scales. For example, interactions between sites less than 100 kilobases apart are bridged by cohesin and Mediator, while those involving sites more than a megabase apart are anchored by CTCF and cohesin or CTCF alone (Phillips-Cremins, Sauria, Sanyal et al. 2013; Bodnar and Spector 2013).
- “Grewal and co-workers found that chromosome arms are organized into locally compacted globules of 50–100 kb in size that require cohesin enrichment at their boundaries. Impairment of cohesin resulted in disruption of these structures and led to loss of chromosome territory restriction and genome-wide transcriptional readthrough. ‘These results reveal that cohesin-dependent globules are basic architectural components of arms and are, in fact, the smallest structural unit yet discovered,’ says Grewal. As for the function of globules on arms, Grewal posits that globules are likely to ‘promote functional annotation of the genome, perhaps by ensuring confinement of RNA polymerase II transcriptional activity’. Surprisingly, heterochromatin provided additional structural constraints at centromeres and telomeres, which in effect shape 3D genome architecture by constraining interactions between chromosome arms” (Koch 2014, doi:10.1038/nrg3858).
-
A new study of lymphoblastoid cells and other human cell lines has — at
unprecedented resolution — revealed yet finer and more complex details
of chromosome organization and dynamics. The authors report chromosome
domains (“contact domains”, because loci within the domain interact
with each other much more frequently than with loci outside the domain)
ranging from 40 kilobases to 3 megabases in size, with a median size of
185 kilobases. These domains often occur as loops (about 10,000 of
them), typically “knotted” by CTCF protein, and about 30% of the time
(in lymphoblastoid cells) the loops bring gene enhancers and promoters
into contact. The domains fall into at least six qualitatively
distinct types (or “flavors”) distinguished by, among other things,
their chromatin modifications and their long-range chromosome contacts.
When the pattern of chromatin modifications changes, the pattern of
long-range contacts also changes. All the domains of a given type —
that is, all their occurrences on all the chromosomes — tend to
colocate in one “compartment” of the nucleus, so that there are at
least six such compartments.
But all this needs to be put in a dynamic context. As the authors summarize the matter in a video abstract of their paper in the journal Cell, “A loop that turns a gene on in one cell type might disappear in another. A domain may move from subcompartment to subcompartment as its flavor changes. No two cell types [have their chromosomes] folded alike. Folding drives function. Epigenetics is origami”. (Rao, Huntley, Durand et al. 2014, doi:10.1016/j.cell.2014.11.021).
The key take-home lesson: “folding drives function”. This is a long way from the old view that “sequence tells us everything we need to know about function”. It’s the difference between stasis, on the one hand (imagine the bits of a computer program, statically embodied in transistors or optical disks), and a physical embodiment that is at the same time a sculptural and balletic performance, on the other hand. In the latter case, the performance governs the functional story. Analogizing this to a computer would require the computer chips to writhe and dance, thereby imparting to the individual bits their functional meaning.
- “Heterogeneous structures exist among TADs, and this structural heterogeneity is significantly correlated to DNA sequences, epigenomic signals and gene expressions. Although TADs can be stable in genomic positions across cell lines, structural comparisons show that a considerable number of stable TADs undergo significantly structural rearrangements during cell changes. Moreover, the structural change of TAD is tightly associated with its transcription remodeling” (Wang, Dong, Zhang and Peng 2015, doi:10.1093/nar/gkv684).
- “The liquid-like movement of chromatin should bring about fluctuation of the TAD structure. A simulation study using a polymer model showed that these domains should fluctuate between open and closed structures. A simulation [of certain data sets] suggested the TADs fluctuate among multiple structures, showing the importance of entropy effects. Correlation between the structural changes of the chromatin domain and the expression level of genes included in that particular domain has been shown by comparing cells with different expression levels” (Maeshima, Ide, Hibino and Sasai 2016, doi:10.1016/j.gde.2015.11.006).
- “Here we show ... that genomic duplications in patient cells and genetically modified mice can result in the formation of new chromatin domains (neo-TADs) and that this process determines their molecular pathology ... Our findings provide evidence that TADs are genomic regulatory units with a high degree of internal stability that can be sculptured by structural genomic variations” (Franke, Ibrahim, Andrey et al. 2016, doi:10.1038/nature19800).
- Regarding the Sonic hedgehog gene (Shh) and its limb enhancer (ZRS): “The formation of a compact topological domain enables the Shh limb enhancer to activate gene expression across very large genomic distances. Although enhancer activity is pervasive throughout the TAD, it is not uniform. Genes located in certain ‘cold spots’ are less affected by the activity of the enhancer, either due to the specific folding of chromatin within the TAD or due to local chromatin effects. When the surrounding TAD is disrupted and made less compact (e.g., by a genomic inversion encompassing one of the TAD boundaries), the activity of the limb enhancer becomes dependent on the genomic distance between the enhancer and a target gene” (Beagrie and Pombo 2016, doi:10.1016/j.devcel.2016.11.011).
-
Chromosome territories
Individual chromosomes tend to occupy particular areas of the nucleus,
which can change with cell type, stage of the cell cycle, and various other
conditions. The areas are not mutually exclusive, as all the kinds of
chromosome interaction testify.
- There is a connection between the organization of chromosomes and disease. “Repositioning of chromosome territories that results from chromosome translocations (often associated with with tumorigenesis) notably affects the transcriptome [products of gene expression], and distinct positional changes are observed during normal and tumorigenic differentiation” (Joffe, Leonhardt and Solovei 2010).
- Researchers at the Nencki Institute of Experimental Biology of the Polish Academy of Sciences have shown that the “memory” of past events by neurons correlates with changed positions of certain genes, for example, in relation to the nuclear membrane. “While conducting experiments on rats after epileptic seizures we have observed that a gene may permanently move deeper into the neuron’s cell nucleus. Since modification of the geometrical structure of the nucleus leads to changes in gene expression, this is how the neuron remembers what happened” (Prof. Grzegorz Wilcyński, quoted in Nencki Institute of Experimental Biology 2013).
- Researchers who triggered the expression of NF-κB (a transcription factor associated with the inflammation response) found that the levels of hundreds of long noncoding RNAs were driven up or down — and 54 of these derived from pseudogenes.
- “Chromosome territories (CTs) in higher eukaryotes occupy tissue-specific non-random three-dimensional positions in the interphase nucleus. To understand the mechanisms underlying CT organization, we mapped CT position and transcriptional changes in undifferentiated embryonic stem (ES) cells, during early onset of mouse ES cell differentiation and in terminally differentiated NIH3T3 cells. We found chromosome intermingling volume to be a reliable CT surface property, which can be used to define CT organization. Our results show a correlation between the transcriptional activity of chromosomes and heterologous chromosome intermingling volumes during differentiation. Furthermore, these regions were enriched in active RNA polymerase and other histone modifications in the differentiated states” (Maharana, Iyer, Jain et al., doi:10.1093/nar/gkw131)
-
Radial positioning of chromosome segments
- General (non-absolute) rule: genes located toward the periphery of the nuclear volume tend to be repressed, while genes located toward the center tend to be active. However, numerous different and conflicting patterns of response are being observed.
- “In spherical nuclei, such as lymphocytes, the radial positioning of chromosomes correlates with their gene density, with gene-dense and gene-poor chromosomes positioned centrally or at the periphery, respectively. In cells with flat nonspherical nuclei, such as fibroblasts, the size of the chromosome correlates with the radial position, with smaller chromosomes occupying central positions of the nucleus and larger chromosomes positioned toward the periphery independently of gene density” (Ferrai, de Castro, Lavitas et al. 2010).
-
Colocalization of genes (and "transcription factories")
“Transcription factories” are foci within the nucleus where trancription
and various factors required for transcription are concentrated. “Several
genomic loci can share a single transcription factory, and in some cases
appear to do so non-randomly, suggesting that factories may physically
coordinate transcription and gene expression inside the nuclear space”.
Colocalized genes may be from the same or different chromosomes. Taken
together, current studies “suggest that the genome within a mammalian cell
nucleus is subject to regulated and tissue-specific three-dimensional
structuring” (Edelman and Fraser 2012).
“Because many fewer Pol II foci were detected [40 to
200 per cell] compared to the number of actively transcribed genes per
nucleus, the factory model proposed that multiple coexpressed genes move in
and out of preassembled factories. With advances in live imaging, we now
know that the system is much more dynamic. For example, super-resolution
live imaging revealed highly dynamic and transient clusters of Pol II.
These clusters do not reside in fixed locations within the nucleus, but are
instead formed de novo upon transcriptional stimulation, persisting for
short periods, on the order of a minute”
(Furlong and Levine 2018, doi:10.1126/science.aau0320).
A dynamic variant of the transcription factory model (hubs) is gaining
momentum as it incorporates features of all classical models of
enhancer-promoter interactions, explains many observations reported for
transcription factories, and accounts for more contemporary observations
such as transcriptional bursting. According to this model, prelooped
topologies serve as hubs or traps for the accumulation of Pol II and other
complexes required for gene expression. Liquid-liquid phase transitions
were proposed to facilitate this process because many TFs, coactivators, and
components of the basal transcription machinery contain intrinsically
disordered domains that can foster such interactions. Studies of the
assembly of germline determinants (P-granules) in Caenorhabiditis
elegans indicate that different RNA and protein subunits associate
through such phase transitions.
(Furlong and Levine 2018, doi:10.1126/science.aau0320).
- “Just as the spatial vicinity of two PREs [DNA sequences known as ‘polycomb response elements’] produces a pairing-dependent enhancement of silencing, the vicinity of two derepressed PcG [polycomb group] target genes might result in enhanced transcriptional activity or stability of the active state. A remarkable report concerning the transcriptional activity of the Drosophila Hox gene Ubx demonstrated in fact that, when two copies of the gene are homologously paired, transcription from each allele is enhanced, while chromosome rearrangements that prevent pairing reduce transcription” (Pirrotta and Li 2012).
- In yeast “a considerable number of transcription factors (TFs) regulate genes that are colocalized in the nucleus. Colocalized TF target genes are more strongly coregulated compared with the other TF target genes. Target genes of chromatin regulators are also colocalized. These results demonstrate that colocalization of coregulated genes is a common process, and three-dimensional gene positioning is an important part of gene regulation” (Dai and Dai 2012).
- Also in yeast, it’s been shown how a transcription factor and various nuclear pore complex proteins can interact with genes so as to localize these genes (which can be from different chromosomes) in a cluster at the nuclear envelope and activate them. At least some of the genes in the same cluster have the same “gene recruitment sequences” in their promoters. There are indications that similar processes occur in multicellular organisms (Burns and Wente 2012). Little is known about how the regulatory proteins coordinate this.
- Experiments show that “the proteomes of each [transcription factory] complex contained hundreds of distinct factors, many previously known to be involved in transcription and specific to that particular transcription complex. Each factory type also shared a suite of general factors” (Edelman and Fraser 2012).
- “The dynamical process of gene co-localization at factories has been shown to depend on the action of specific transcriptional regulatory factors, and once there, genes are ‘tethered’ at the position of the progressing [RNA] polymerase” (Edelman and Fraser 2012).
- As in virtually all things molecular biological, the story only becomes more complex with further investigation: A group of researchers “show that transcription factories operate in mammalian cells and that transcription initiation and elongation steps take place in different compartments. Their findings suggest a modified transcription factory model whereby genes form nuclear foci at the initiation step, but transcription moves out of these foci during elongation” (doi:10.1101/gad.216200.113).
- Questions: “How do the multitude of factors that make up factories assemble into foci as cells exit from mitosis? Do promoters and enhancers of ‘active genes to be’ serve as binding scaffolds that then coalesce into transcription foci in the first hour of G1 when chromatin is known to be highly dynamic? And what of genes switched on during interphase, how do they move to these sites and what mechanisms govern their preferential organization?” (Edelman and Fraser 2012).
-
Chromosomal rearrangements
- Increasing knowledge of genome integrity and folding, together with investigations of genetic recombination, “strongly suggests that partner choice in chromosomal rearrangement primarily follows the three-dimensional conformation of the genome”. This rearrangement can be a major factor in cancers, and is also common in normal cells (Wijchers and de Laat 2011).
-
Nuclear matrix
The nuclear matrix is an ill-defined network of fibres in the nucleus.
Not much is yet known about its functions. However, there are fairly well
characterized “matrix attachment regions” (MARs — sometimes referred to as
scaffold/matrix attachment regions, or S/MARs) occurring along the length
of chromsomes. These are thought to be targeted by regulatory proteins
that play a role in “attaching” particular chromosome segments to various
nuclear compartments, such as so-called “transcription factories”.
- Matrix attachment regions (MARs) of chromosomes seem to help regulate miRNAs, which in turn are major regulators of gene expression (see “MicroRNA (miRNA) activity” above). Recent work “implies that the association of MAR binding proteins to MARs could dictate the tissue/context specific regulation of miRNA genes” (Chavali, Funa and Chavali 2011).
-
Nuclear envelope
“The nuclear periphery has conventionally been considered as a
zone of inactive chromatin and transcriptional repression. Recent
studies ... reveal a complex picture. Whilst the edge of the
nucleus does seem to have a direct effect on the expression of
some genes, other genes seem unaffected by their proximity to the
nuclear periphery. Moreover, the nuclear periphery itself is
heterogeneous, with microdomains of differing compositions,
associating with different genomic regions and probably having
differential effects on genome function” (Deniaud and Bickmore
2009). There is, in other words “a complex heterogeneity at the nuclear
periphery” supporting “crosstalk between genes, genetic elements,
perinuclear compartments and the nuclear envelope” (Mekhail and Moazed
2010).
“The nuclear lamina and the NPC [nuclear pore complex] both contribute to
the organization of the genome and to transcription regulation. Whereas the
net effect of lamina association is almost always gene repression,
association with NPCs can either activate or repress genes. Association of a
locus with the lamina does not induce its association with nearby NPCs,
underscoring the independence of these modes of tethering. The factors
that dictate whether NPC–genome interactions activate or repress loci remain
to be determined. Moreover, it remains unclear how individual lamin isoforms
and lamina-associated proteins contribute to scaffolding LADs
[lamina-associated domains]. However, it is evident that lamina association
is only one of several strategies for repressing gene activity, as of the
thousands of genes that decrease in expression during ES [embryonic stem]
cell differentiation, only hundreds become LAD residents.
It may be that nuclear lamina-directed genome organization is more important in differentiated cell types, as pluripotent cells lacking B-type lamins exhibit surprisingly modest defects in genome organization. Compared with LADs, NUP-binding profiles are considerably narrower and cover a smaller and more variable portion of the genome. NUPs often bind at the level of individual regulatory elements associated with a specific gene, including enhancers, promoters and transcription start sites. Unlike LADs, NUP [nucleoporin protein] interactions can be rapidly induced or disrupted by environmental stimuli.
Clearly, cells balance several strategies for organizing genomes ...” (Buchwalter, Kaneshiro and Hetzer 2018, doi:10.1038/s41576-018-0063-5)
“The nuclear compartment is delimited by a specialized expanded sheet of the
endoplasmic reticulum (ER) known as the nuclear envelope (NE). Compared to
the outer nuclear membrane and the contiguous peripheral ER, the inner
nuclear membrane (INM) houses a unique set of transmembrane proteins that
serve a staggering range of functions. Many of these functions reflect the
exceptional position of INM proteins at the membrane–chromatin interface.
Recent research revealed that numerous INM proteins perform crucial roles in
chromatin organization, regulation of gene expression, genome stability, and
mediation of signaling pathways into the nucleus. Other INM proteins
establish mechanical links between chromatin and the cytoskeleton, help NE
remodeling, or contribute to the surveillance of NE integrity and
homeostasis”
(Pawar and Kutay 2021, doi:10.1101/cshperspect.a040477).
-
Lamins and lamin-binding proteins
“Nuclear lamins are ancient type V intermediate filaments with diverse
functions that include maintaining nuclear shape, mechanosignaling,
tethering and stabilizing chromatin, regulating gene expression, and
contributing to cell cycle progression ... Accumulating work indicates
that a range of lamin post-translational modifications (PTMs) control
their functions both in homeostatic cells and in disease states such as
progeria, muscular dystrophy, and viral infection”. “Two classes of
lamins form overlapping networks at the nuclear periphery but have
distinct regulatory roles”. Relevant post-translational modifications
include phosphorylation, acetylation, ubiquitination, SUMOylation,
methylation, and O-GlnAcylation. “While phosphorylation is a known
regulator of mitotic laminar disruption, diverse post-translational
modifications are being established as toggles of lamin arrangement,
interactions, and functions”
(Murray-Nerger and Cristea 2021, doi:10.1016/j.tibs.2021.05.007).
The nuclear lamina is a filamentous protein network lining the inner
nuclear membrane. “Through interactions with cytoplasmic and nuclear
components, the nuclear lamina defines the shape and mechanical
properties of the nucleus. Together with other inner nuclear membrane
proteins, lamins also tether transcription factors and signaling
molecules. As such, lamins can be perceived as a relay platform for
intracellular signaling pathways reaching the nuclear interior. ...
Lamins also interact with chromatin. The role of the nuclear envelope
and nuclear lamins on the spatial arrangement of chromosomes and
epigenetic modifications suggests a tight interplay between lamins,
chromatin organization, and gene expression” (Collas, Lund and
Oldenburg 2014).
“Lamin-binding proteins appear to serve as the ‘adaptors’ by which the
lamina organizes chromatin, influences gene expression and epigenetic
regulation, and modulates signaling pathways. Transient interactions
of lamins with key components of the transcription and replication
machinery may provide an additional level of regulation or support to
these essential events” (Wilson and Foisner 2010).
There are two types of lamins. “Whereas B-type lamins are ubiquitously
expressed, A-type lamins are developmentally regulated: they are absent
from early embryos, but are expressed in lineage-committed progenitor
cells and in differentiated cells, with however some exceptions. A- and
B-type lamins are post-translationally processed differently” (Collas,
Lund and Oldenburg 2014).
“Recent studies ... provide evidence that spatiotemporal differences in
lamina composition and genome architecture underlie developmental
competence and differentiation, suggesting the nuclear lamina is
directly involved in spinning the web of cell fate” (Van Bortle and
Corces 2013).
“The nuclear lamina (NL) is a protein scaffold lining the nuclear
envelope that consists of nuclear lamins and associated transmembrane
proteins. It helps to organize the nuclear envelope, chromosomes, and the
cytoplasmic cytoskeleton. The NL also has an important role in regulation
of signaling, as highlighted by the wide range of human diseases caused
by mutations in the genes for NL proteins with associated signaling
defects. This review will consider diverse mechanisms for signaling
regulation by the NL that have been uncovered recently, including
interaction with signaling effectors, modulation of actin assembly and
compositional alteration of the NL. Cells with discrete NL mutations
often show disruption of multiple signaling pathways, however, and for
the most part the mechanistic basis for these complex phenotypes remains
to be elucidated”
(Gerace and Tapia 2018, doi:10.1016/j.ceb.2017.12.009).
- “Molecular mapping studies have now identified large genomic domains that are in contact with the lamina. Genes in these domains are typically repressed, and artificial tethering experiments indicate that the lamina can actively contribute to this repression. ... A variety of DNA-binding and chromatin proteins may anchor specific loci to the lamina, while histone-modifying enzymes partly mediate the local repressive effect of the lamina” (Kind and van Steensel 2010).
- “The lamina indirectly controls gene expression in the nuclear interior by sequestration of certain transcription factors” (Kind and van Steensel 2010).
- “Molecular mapping studies [have] indicated that lamina-genome interactions are dynamic and play essential roles in the regulation of gene expression programs during lineage commitment and terminal differentiation” (Li and Reinberg 2011).
- “In adipose stem cells, lamin A associates with thousands of promoters in a manner that modulates transcription outcomes. Lamin A-enriched promoters cluster non-randomly into “lamin-rich domains” ... Whereas the vast majority of these lamin A-enriched genes are not expressed (and are accordingly marked by repressive histone modifications), a small proportion (5%) is marked by H3K4me3 and displays a wide range of expression patterns, some of which being strongly expressed. This suggests that an additional component modulates expression of lamin A-associated genes”. “We found that lamin A tends to occupy distinct promoter sub-regions ... Further, the position of a lamin A peak on a promoter region correlates with a given gene expression outcome: lamin A interaction with proximal promoter regions or at the TSS [transcription start site] and downstream strongly correlates with gene inactivity, even when the TSS is enriched in the transcriptionally permissive H3K4me3 mark. In contrast, lamin A association with a more upstream distal region is permissive, that is, compatible with gene expression if H3K4me3 marks the TSS. These findings are consistent with a view of lamin A association with the TSS area hindering nucleosome turnover associated with gene activation, access to chromatin remodelers or recruitment of the transcription machinery” (Collas, Lund and Oldenburg 2014).
- “In an extreme twist on the relationship between nuclear organization and genome function, specific cell types exhibit an ‘inside-out’ architecture in which genes and markers of active chromatin are found exclusively at the nuclear periphery and heterochromatin centrally positioned. Nuclear inversion occurs in the nuclei of mouse retinal rod cells, wherein rearrangement of chromatin takes place during terminal differentiation of rod nuclei and affects the optical properties of the retina by reducing light scattering in the outer nuclear layer. This unusual pattern of nuclear inversion also develops in the rod nuclei of several other nocturnal mammals, suggesting rearrangement of chromatin represents an adaptation for night vision. Nuclear inversion is gradually established over several weeks, and a recent follow up study suggests that changes in nuclear lamina composition underlie the dynamic arrangement and maintenance of chromatin organization in the differentiating rod cells” (Van Bortle and Corces 2013).
- “This review highlights a prominent family of nuclear lamina proteins that carries the LAP2-emerin-MAN1-domain (LEM-D). LEM-D proteins share an ability to bind lamins and tether repressive chromatin at the nuclear periphery. The importance of this family is underscored by findings that loss of individual LEM-D proteins causes progressive, tissue-restricted diseases, known as laminopathies. Diverse functions of LEM-D proteins are linked to interactions with unique and overlapping partners including signal transduction effectors, transcription factors and architectural proteins. Recent investigations suggest that LEM-D proteins form hubs within the nuclear lamina that integrate external signals important for tissue homeostasis and maintenance of progenitor cell populations” (Barton, Soshnev and Geyer 2015, doi:10.1016/j.ceb.2015.03.005).
-
Cytoskeleton-DNA bridges
- There are protein-protein interactions through the nuclear envelope that link the cytoskeleton (in the cell cytoplasm) to chromatin in the nuclear interior. These links “are crucial to the balancing of subcellular forces as well as to the maintenance of nuclear structure and overall genome integrity” (Mekhail and Moazed 2010).
-
Nuclear pore complexes
“In eukaryotic cells, the genetic material is segregated inside the
nucleus. This compartmentalization of the genome requires a transport
system that allows cells to move molecules across the nuclear envelope,
the membrane-based barrier that surrounds the chromosomes. Nuclear pore
complexes (NPCs) are the central component of the nuclear transport
machinery. These large protein channels penetrate the nuclear envelope,
creating a passage between the nucleus and the cytoplasm through which
nucleocytoplasmic molecule exchange occurs. NPCs are one of the largest
protein assemblies of eukaryotic cells and, in addition to their critical
function in nuclear transport, these structures also play key roles in
many cellular processes in a transport-independent manner. Here we will
review the current knowledge of the NPC structure, the cellular
mechanisms that regulate their formation and maintenance, and we will
provide a brief description of a variety of processes that NPCs regulate”
(Raices and D’Angelo 2022, doi:10.1101/cshperspect.a040691).
“Today nuclear pores are not anymore seen as static facilitators of
nucleocytoplasmic transport but ensembles of multiple overlaying
functional states that are involved in various cellular processes” (Hurt
and Beck 2015, https://dx.doi.org/10.1016/j.ceb.2015.04.009).
“Accumulating evidence shows that besides their main role in regulating
the exchange of molecules between these two compartments [nucleus and
cytoplasm], NPCs and their components also play important
transport-independent roles, including gene expression regulation,
chromatin organization, DNA repair, RNA processing and quality control,
and cell cycle control”
(Raices and D’Angelo 2017, doi:10.1016/j.ceb.2016.12.006).
Nuclear pore complexes “form highly dynamic ensembles. Nuclear pore
composition is not only variable across the tree of life, but also across
healthy and disease tissues in multicellular organisms, during
reprogramming and cell differentiation and possibly even within single
cells. Nuclear pore structure and function might be further modulated by
conformational heterogeneity and post-translational modifications. We are
just beginning to understand the functional consequences of such
structural variants” (Hurt and Beck 2015, doi:10.1016/j.ceb.2015.04.009).
“Mounting evidence has implicated a group of proteins termed
nucleoporins, or Nups, in various processes that regulate chromatin
structure and function. Nups were first recognized as building blocks for
nuclear pore complexes, but several members of this group of proteins
also reside in the cytoplasm and within the nucleus. Moreover, many are
dynamic and move between these various locations. Both at the nuclear
envelope, as part of nuclear pore complexes, and within the nucleoplasm,
Nups interact with protein complexes that function in gene transcription,
chromatin remodeling, DNA repair, and DNA replication”
(Ptak and Wozniak 2016, doi:10.1016/j.ceb.2016.03.024).
“Regulation of gene expression at the pore can be governed through
tethering of genomic regions to the pore via interactions with
nucleoporins and transcription factors. This can lead to either induction
or repression of transcriptional activity of genes repositioned at the
nuclear periphery”
(Ben-Yishay, Ashkenazy and Shav-Tal 2016, doi:10.1016/j.tig.2016.04.003).
“Many chromosomal loci physically interact with nuclear pore proteins
(Nups), and interactions with Nups can promote transcriptional
repression, transcriptional activation, and transcriptional poising.
Interaction with the NPC also affects the spatial arrangement of genes,
interchromosomal clustering, and folding of topologically associated
domains. Thus, the NPC is a spatial organizer of the genome and regulator
of genome function”
(Sumner and Brickner 2022, doi:10.1101/cshperspect.a039438).
- “Although the nuclear pore complex (NPC) is best known for its primary function as the key regulator of molecular traffic between the cytoplasm and the nucleus ... the NPC is emerging as an important regulator of gene expression through its influence on the internal architectural organization of the nucleus and its apparently extensive involvement in coordinating the seamless delivery of genetic information to the cytoplasmic protein synthesis machinery” (Strambio-De-Castillia, Niepel and Rout 2010). “Recent data suggest a view of the NPC as one of the central hubs at which crucial gene regulatory pathways converge. Import of transcription factors, assembly of transcription machinery and distant gene loci, mRNA processing and export processes, and finally, the memory of these activating events through cell divisions may all in some ways [be] connected to the NPC” (Liang and Hetzer 2011).
- Knockdown of an NPC protein (NUP210) that is integral to the nuclear membrane results in failure of at least two cellular differentiation processes in mouse embryos. In one cell type, 64 genes were upregulated and 191 were downregulated. “As the NPC is viewed as a site of transcription, one possible mechanism is that NUP210 modulates the activity of transcriptional regulatory proteins” (Muers 2012, reporting on a paper by M. A. D’Angelo et al. doi:10.1016/j.devcell.2011.11.021).
- There are DNA sequences (so-called “zip codes”) associated with some gene promoters. These, in conjunction with certain components of the nuclear pore complex (and presumably also with a role for transcription factors) effectively target the gene to the nuclear periphery. Full activation of such genes can depend on their being located at the periphery, and therefore depends on the presence of the NPC components. The zip codes point to “an additional level of genetic information that controls the spatial organization of the genome and affects gene expression” (Ahmed, Brickner, Light et al. 2010).
- “The interaction of nuclear pore proteins (Nups) with active genes can promote their transcription. In yeast, some inducible genes interact with the nuclear pore complex both when active and for several generations after being repressed, a phenomenon called epigenetic transcriptional memory. This interaction promotes future reactivation and requires Nup100, a homologue of human Nup98. A similar phenomenon occurs in human cells ... In both yeast and human cells, the recently expressed promoters of genes with memory exhibit persistent dimethylation of histone H3 lysine 4 (H3K4me2) and physically interact with Nups and a poised form of RNA polymerase II ... In human cells transiently depleted of Nup98 or yeast cells lacking Nup100, transcriptional memory is lost; RNA polymerase II does not remain associated with promoters, H3K4me2 is lost, and the rate of transcriptional reactivation is reduced (Light, Freaney, Sood et al. 2015, doi:10.1371/journal.pbio.1001524).
- “Here, we show that nuclear pore complex (NPC) components Nup93 and Nup153 bind superenhancers, regulatory structures that drive the expression of key genes that specify cell identity. We found that nucleoporin-associated superenhancers localize preferentially to the nuclear periphery, and absence of Nup153 and Nup93 results in dramatic transcriptional changes of superenhancer-associated genes. Our results reveal a crucial role of NPC components in the regulation of cell type-specifying genes and highlight nuclear architecture as a regulatory layer of genome functions in cell fate” (Ibarra, Benner, Tyagi et al. 2016, doi:10.1101/gad.287417.116).
- “We show that the addition of the Nup210 nucleoporin to NPCs during myoblast differentiation results in assembly of an Mef2C transcriptional complex required for efficient expression of muscle structural genes and microRNAs. We show that this NPC-localized complex is essential for muscle growth, myofiber maturation, and muscle cell survival and that alterations in its activity result in muscle degeneration. Our findings suggest that NPCs regulate the activity of functional gene groups by acting as scaffolds that promote the local assembly of tissue-specific transcription complexes and show how nuclear pore composition changes can be exploited to regulate gene expression at the nuclear periphery” (Raices, Bukata, Sakuma 2017, doi:10.1016/j.devcel.2017.05.007).
- “We show that rat heart muscle cells (cardiomyocytes) undergo a 63% decrease in nuclear pore numbers during maturation, and this changes their responses to extracellular signals. The maturation-associated decline in nuclear pore numbers is associated with lower nuclear import of signaling proteins such as mitogen-activated protein kinase (MAPK). Experimental reduction of nuclear pore numbers decreased nuclear import of signaling proteins, resulting in decreased expression of immediate-early genes. In a mouse model of high blood pressure, reduction of nuclear pore numbers improved adverse heart remodeling and reduced progression to lethal heart failure. The decrease in nuclear pore numbers in cardiomyocyte maturation and resulting functional changes demonstrate how terminally differentiated cells permanently alter their handling of information flux across the nuclear envelope and, with that, their behavior” (Han, Mich-Basso, Li et al. 2022, doi:10.1016/j.devcel.2022.09.017).
-
Lamins and lamin-binding proteins
-
Cell surface
-
Cell adhesion
- Within minutes or even seconds of contacting a foreign surface, including the surface of adjacent cells, the behavior of a cell changes. The chemistry, topography, and mechanical properties of the contacted surface affect “nearly all aspects of cell behavior observed during the days following adhesion”. Further, “long term cell behavior is highly dependent on the alterations of cell shape and cytoskeletal organization that are often initiated during the minutes to hours following adhesion”. The expression of many genes is affected by these processes, but “relating substratum properties to alterations of the expression of hundreds of genes as a consequence of the perturbation of a complex network of biochemical reactions seems a formidable task” (Cretel, Pierres, Benoliel and Bongrand 2008).
-
Endocytosis
Endocytosis is a process by which the cell (plasma) membrane
invaginates and “ingests” substances from outside the cell, forms a
separate container, or vesicle, containing the substances, after which
the vesicle detaches from the membrane and transports the substances
into the interior of the cell.
- “There is growing support for the idea that transcription is directly controlled by endocytic proteins...The regulation of transcription by endocytic proteins occurs at several levels: remodelling of chromatin, regulation of transcription initiation and delivery of transcriptionally relevant cargo”. “It now seems that endocytosis is a master organizer of signalling circuits, with one of its main roles being the resolution of signals in space and time. Many of the functions of endocytosis that are emerging from recent research ... point to endocytosis being integrated at a deeper level in the cellular ‘master plan’ (the cellular network of signalling circuits that lie at the base of the cell’s make-up)” (Scita and Di Fiore 2010).
-
Cell adhesion
-
Cell shape, extracellular matrix, and environment in general
- Changes in cell shape, whether induced by neighboring cells, the extracellular matrix, or applied forces from the environment can result in metabolic changes and transformation of the entire cell phenotype. Changes in gene expression are integral to this transformation. In the case of cancer cells, such external influences have been followed by a reversion to a previous morphology and phenotype. In general, “cell shape is known to influence several gene-regulatory pathways through architectural rearrangement” (D’Anselmi, Valerio, Cucina et al. 2010).
-
Structural proteins
“CTCF and cohesin are essential for genome folding, with their roles in
the formation of topologically associating domains (TADs) particularly
well studied by population-level analyses. Now, a single-cell study
published in Molecular Cell reveals that CTCF and cohesin
contribute to genome organization across multiple scales owing to their
essential roles in the formation and stacking of chromatin loops” (Clyde
2023; doi:10.1038/s41576-023-00616-7).
-
Insulator protein CTCF (CCCTC-binding factor)
Originally described as a transcription factor, CTCF “has been
implicated in diverse cellular processes, including transcriptional
regulation, alternative splicing, insulation, imprinting, X-chromosome
inactivation, and higher-order chromatin organization” (Saldaña-Meyer,
González-Buendia, Guerrero et al. 2014).
- This protein is heavily involved in insulator function. (See “Insulators” under PRE-TRANSCRIPTIONAL DECISION-MAKING above.) There are an estimated 15,000 CTCF binding sites in the genome (Kim, Abdullaev, Smith et al. 2007; Xie, Mikkelsen, Gnirke et al. 2007).
- As an example of transcriptional repression, CTCF binds in the middle of the PUMA (“p53 up-regulated modulator of apoptosis”) gene, blocking transcription by RNA polymerase II. The CTCF binding site marks a boundary between activating and repressive histone modifications. It appears that the p53 “tumor-suppressor” protein can disrupt the CTCF binding and allow transcription of PUMA (Gomes and Espinosa 2010).
- “It has ... been recently suggested that the primary function of CTCF binding may be in establishing the three-dimensional organization of the genome within the nucleus and that potential regulatory effects of CTCF binding may be secondary to this principal role. Consistent with this idea, CTCF mediates chromatin looping at a number of loci” (Noonan and McCallion 2010).
- Actually, the involvement of CTCF in such three-dimensional aspects of the nucleus as chromosome looping and the bringing together of enhancers and promoters is now well-established and known to be widespread, although the way all this is accomplished remains unknown (Krivega and Dean 2012; Yang and Corces 2012). See also “Insulators” under PRE-TRANSCRIPTIONAL DECISION-MAKING
- “Because of its critical role in genome function, CTCF binding patterns have long been assumed to be largely invariant across different cellular environments”. However, recent observations show "surprisingly plastic [CTCF] genomic binding landscapes, indicative of strong cell-selective regulation of CTCF occupancy”. Some “41% of variable CTCF binding is linked to differential DNA methylation, concentrated at two critical positions within the CTCF recognition sequence”, but 36% of variable CTCF binding was independent of methylation and “may derive from complex regulation of co-factors or variation in its specific interaction partners. Given the breadth of CTCF’s regulatory functionality, our observation of global binding variation implies a widespread potential role in the translation of epigenetic marks to genome organization at thousands of sites” (Wang, Maurano, Qu et al. 2012).
- “Activation of retroelements [retrotransposons] has produced species-specific expansions of CTCF binding in rodents, dogs, and opossum”. Over 5000 binding sites for CTCF were found to be shared among five representative placental mammals, and many such sites function as barriers separating transcriptionally active from inactive chromatin (Schmidt, Schwalie, Wilson et al. 2012).
- “[We] show that the location and relative orientations of CBSs [CTCF-binding sites] determine the specificity of long-range chromatin looping in mammalian genomes, using protocadherin (Pcdh) and β-globin as model genes. Inversion of CBS elements within the Pcdh enhancer reconfigures the topology of chromatin loops between the distal enhancer and target promoters and alters gene-expression patterns. Thus ... the orientation of at least some enhancers carrying CBSs can determine both the architecture of topological chromatin domains and enhancer/promoter specificity” (Guo, Xu, Canzio et al. 2015, doi:10.1016/j.cell.2015.07.038).
- “Architectural proteins, including CTCF, are the determinants for the strength of TAD [topologically associating domain of DNA] formation and insulator function. The selection of interacting regions, in the case of CTCF, is dictated by the binding site orientation. It is obvious that CTCF and its orientation only partly account for the determinants selecting and mediating proper interactions. About 30,000 sites in the vertebrate genome are bound by CTCF, but only a fraction is found at TAD borders. What are the factors or combinations of factors determining the specificity of interacting elements? Furthermore, not all TAD borders have CTCF sites. Which factors or features are mediating the boundary function in these cases?” (Ali, Renkawitz and Bartkuhn 2016, doi:10.1016/j.gde.2015.11.009).
- “Enhancer regions and transcription start sites of estrogen-target regulated genes are connected by means of Estrogen Receptor (ER) long-range chromatin interactions. Yet, the complete molecular mechanisms controlling the transcriptional output of engaged enhancers and subsequent activation of coding genes remain elusive. Here, we report that CTCF binding to enhancer RNAs is enriched when breast cancer cells are stimulated with estrogen. CTCF binding to enhancer regions results in modulation of estrogen-induced gene transcription by preventing Estrogen Receptor chromatin binding and by hindering the formation of additional enhancer-promoter ER looping. Furthermore, the depletion of CTCF facilitates the expression of target genes associated with cell division and increases the rate of breast cancer cell proliferation. We have also uncovered a genomic network connecting loci enriched in cell cycle regulator genes to nuclear lamina that mediates the CTCF function. The nuclear lamina and chromatin interactions are regulated by estrogen-ER. We have observed that the chromatin loops formed when cells are treated with estrogen establish contacts with the nuclear lamina. Once there, the portion of CTCF associated with the nuclear lamina interacts with enhancer regions, limiting the formation of ER loops and the induction of genes present in the loop. Collectively, our results reveal an important, unanticipated interplay between CTCF and nuclear lamina to control the transcription of ER target genes” (Elisa Fiorito, Sharma, Gilfillan et al. 2016, doi:10.1093/nar/gkw785).
- “Here we examined in vivo an ~80 kb sub-TAD, containing the mouse α-globin gene cluster, lying within a ~1 Mb TAD. “We find that the sub-TAD is flanked by predominantly convergent CTCF-cohesin sites that are ubiquitously bound by CTCF but only interact during erythropoiesis, defining a self-interacting erythroid compartment. Whereas the α-globin regulatory elements normally act solely on promoters downstream of the enhancers, removal of a conserved upstream CTCF-cohesin boundary extends the sub-TAD to adjacent upstream CTCF-cohesin-binding sites. The α-globin enhancers now interact with the flanking chromatin, upregulating expression of genes within this extended sub-TAD. Rather than acting solely as a barrier to chromatin modification, CTCF-cohesin boundaries in this sub-TAD delimit the region of chromatin to which enhancers have access and within which they interact with receptive promoters” (Hanssen, Kassouf, Oudelaar et al. 2017, doi:10.1038/ncb3573). In sum, “CTCF–cohesin binding sites at the α-globin gene cluster function as boundaries to restrict the interaction of enhancers with the flanking chromatin, thus preventing abnormal gene expression” (table of contents blurb).
- “In eukaryotes, genomic DNA is extruded into loops by cohesin. By restraining this process, the DNA-binding protein CCCTC-binding factor (CTCF) generates topologically associating domains (TADs) that have important roles in gene regulation and recombination during development and disease. How CTCF establishes TAD boundaries and to what extent these are permeable to cohesin is unclear. Here, to address these questions, we visualize interactions of single CTCF and cohesin molecules on DNA in vitro. We show that CTCF is sufficient to block diffusing cohesin, possibly reflecting how cohesive cohesin accumulates at TAD boundaries, and is also sufficient to block loop-extruding cohesin, reflecting how CTCF establishes TAD boundaries. CTCF functions asymmetrically, as predicted; however, CTCF is dependent on DNA tension. Moreover, CTCF regulates cohesin’s loop-extrusion activity by changing its direction and by inducing loop shrinkage. Our data indicate that CTCF is not, as previously assumed, simply a barrier to cohesin-mediated loop extrusion but is an active regulator of this process, whereby the permeability of TAD boundaries can be modulated by DNA tension. These results reveal mechanistic principles of how CTCF controls loop extrusion and genome architecture” (Davidson, Barth, Zakzek et al. 2023, doi:10.1038/s41586-023-05961-5).
-
Cohesin
Cohesin is best known as the protein that, during cell division and
after chromosome replication, holds the sister chromatids together
until they are segregated. Study of the role of cohesin in gene
regulation is only at its beginning, but it has been shown to be
necessary for the expression of a number of genes in different
organisms. “The cohesin complex has recently been shown to be a key
regulator of eukaryotic gene expression, although the mechanisms by
which it exerts its effects are poorly understood” (Liu, Zhang, Bando
et al. 2010; see also Seitan and Merkenschlager 2012).
“The cohesin complex has an essential role in maintaining genome
organization. However, its role in gene regulation remains largely
unresolved. Here we report that the cohesin release factor WAPL creates a
pool of free cohesin, in a process known as cohesin turnover, which
reloads it to cell-type-specific binding sites. Paradoxically,
stabilization of cohesin binding, following WAPL ablation, results in
depletion of cohesin from these cell-type-specific regions, loss of gene
expression and differentiation. Chromosome conformation capture
experiments show that cohesin turnover is
important for maintaining promoter–enhancer loops. Binding of
cohesin to cell-type-specific sites is dependent on the pioneer
transcription factors OCT4 (POU5F1) and SOX2, but not NANOG. We show the importance of cohesin turnover in
controlling transcription and propose that a cycle of cohesin loading and
off-loading, instead of static cohesin binding, mediates promoter and
enhancer interactions critical for gene regulation”
(Liu, Maresca, Brand et al. 2021, doi:10.1038/s41588-020-00744-4).
“Beyond its originally discovered role tethering replicated sister
chromatids, cohesin has emerged as a master regulator of gene expression.
Recent advances in chromatin topology resolution and single-cell studies
have revealed that cohesin has a pivotal role regulating highly dynamic
chromatin interactions linked to transcription control. The dynamic
association of cohesin with chromatin and its capacity to perform loop
extrusion contribute to the heterogeneity of chromatin contacts.
Additionally, different cohesin subcomplexes, with specific properties
and regulation, control gene expression across the cell cycle and during
developmental cell commitment. Here, we discuss the most recent
literature in the field to highlight the role of cohesin in gene
expression regulation during transcriptional shifts and its relationship
with human diseases”
(Perea-Resa, Wattendorf, Marzouk and Blower 2021,
doi:10.1016/j.tcb.2021.03.005).
- Cohesin often works in conjunction with CTCF (see above); however, “while cohesin is recruited by CTCF, local chromosome conformation is defined by cohesin, not CTCF” (Merkenschlager 2010). And, in fact, it is now known that cohesin localizes to DNA regions that, while lacking CTCF binding sites, contain binding sites for known, tissue-specific regulators of gene expression (Ohlsson 2010).
- “We show that very specific long-range interactions are anchored by cohesin/CTCF sites, but not cohesin-only or CTCF-only sites, to form a hierarchy of chromosomal loops”. This hierarchy of long-range interactions is “remarkably selective and complex”. Further, this looping has the effect, not only of bringing sites together, but also of insulating particular regions against contact from each other (Sofueva, Yaffe, Chan et al. 2013).
- Cohesin interacts directly with Mediator, a major part of the Pre-Initiation Complex required for transcription of protein-coding genes. “The Mediator-cohesin complexes promote and/or stabilize the physical proximity between enhancers and promoters of active genes” — but not of inactive ones. (Ohlsson 2010, citing work by M. H. Kagey et al. Nature vol. 467, pp. 430-5 (2010))
- Cohesin cooperates with Polycomb group proteins to achieve partial gene repression (Dorsett 2011).
- In the yeast, Schizosaccharomyces pombe, “small domains of chromatin interact locally on chromosome arms to form globules, which depend on cohesin but not heterochromatin for formation, and heterochromatin at centromeres and telomeres provides crucial structural constraints to shape genome architecture” (blurb in Nature for doi:10.1038/nature13833). The authors write that “Together, our analyses uncover fundamental genome folding principles that drive higher-order chromosome organization crucial for coordinating nuclear functions”.
- “Cohesin can diffuse rapidly on DNA in a manner consistent with topological entrapment and can pass over some DNA‐bound proteins and nucleosomes but is constrained in its movement by transcription and DNA‐bound CCCTC‐binding factor (CTCF). These results indicate that cohesin can be positioned in the genome by moving along DNA, that transcription can provide directionality to these movements, that CTCF functions as a boundary element for moving cohesin, and they are consistent with the hypothesis that cohesin spatially organizes the genome via loop extrusion” (Davidson, Goetz, Zaczek et al. 2016, doi:10.15252/embj.201695402).
- “The cohesin core complex possesses an intrinsic ability to traverse DNA in an adenosine triphosphatase (ATPase)‐dependent manner. Translocation ability is suppressed in the presence of Wapl‐Pds5 and Sororin; this suppression is alleviated by the acetylation of cohesin and the action of mitotic kinases. In Xenopus laevis egg extracts, cohesin is translocated on unreplicated DNA in an ATPase‐ and Smc3 acetylation‐dependent manner. Cohesin movement changes from bidirectional to unidirectional when cohesin faces DNA replication; otherwise, it is incorporated into replicating DNA without being translocated or is dissociated from replicating DNA” (Kanke, Tahara, Veld and Nishiyama 2016, doi:10.15252/embj.201695756).
-
Condensin
“Although condensin protein complexes have long been known for their
central role during the formation of mitotic chromosomes, new evidence
suggests they also act as global regulators of genome topology during all
phases of the cell cycle. By controlling intra-chromosomal and
inter-chromosomal DNA interactions, condensins function in various
contexts of chromosome biology, from the regulation of transcription to
the unpairing of homologous chromosomes” (Frosi and Haering 2015,
doi:10.1016/j.ceb.2015.05.008).
- In T cells (part of the immune system), condensin helps effect a “specialized form of chromatin compaction that regulates access of transcription factors to their target loci and so maintains quiescence in the immune system” (Wood and Bickmore 2011). This quiescence ends when the T cells receive a certain stimulation and are presented with appropriate antigens.
- Other roles for gene regulation by condensin are beginning to come into view. “Intriguingly, mammalian erythroid [red blood cell] maturation is associated with chromatin condensation prior to enucleation [elimination of the nucleus, which mature red blood cells lack], and the condensin II subunit CAP-G2 has been implicated in repressing transcriptional activation and promoting terminal differention of erythroid cells” (Wood and Bickmore 2011).
-
Actin
Actin polymers form part of the cytoskeleton in the cytoplasm of
eukaryotic cells. Actin has also been found in the nucleus, but in
less obvious structures, and with uncertain function. However, some
things have now been learned about its role:
“Monomeric actin seems to be involved in regulation of gene expression
through transcription factors, chromatin regulating complexes and RNA
polymerases. In addition to cytoplasmic actin regulators, nuclear
proteins, such as emerin, can regulate actin polymerization properties
specifically in this compartment. Besides of structural roles, nuclear
actin filaments may be required for organizing the nuclear contents and
for the maintenance of genomic integrity”
(Virtanen and Vartiainen 2017, doi:10.1016/j.ceb.2016.12.004).
- “Actin is essential for transcription from RNA polymerases I, II and III. In Pol I transcription, actin and myosin (MYO1C, which binds DNA) act as a molecular motor. For Pol II transcription, β-actin is needed for the formation of the pre-initiation complex. Pol III contains β-actin as a subunit. Actin can also be a component of chromatin remodeling complexes as well as pre-mRNP particles (that is, precursor messenger RNA bundled in proteins), and is involved in nuclear export of RNAs and proteins” (Wikipedia entry, “Actin”).
- A link has been found “between actin and its related or binding proteins and siRNA-mediated transcriptional gene silencing”. The actin cytoskeleton seems to be associated with the delivery of gene silencing components to the nucleus (Ahlenstiel, Lim, Cooper et al. 2012).
-
Microtubules
“Microtubules are major cytoskeletal components mediating fundamental
cellular processes, including cell division. Recent evidence suggests
that microtubules also regulate the nucleus during the cell cycle’s
interphase stage. Deciphering such roles of microtubules should uncover
direct crosstalk between the nucleus and cytoplasm, impacting genome
function and organismal health. Here, we review emerging roles for
microtubules in interphase genome regulation. We explore how microtubules
exert cytoplasmic forces on the nucleus or transport molecular cargo,
including DNA, into or within the nucleus. We also describe how
microtubules perform these functions by establishing transient or stable
connections with nuclear envelope elements. Lastly, we discuss how the
regulation of the nucleus by microtubules impacts genome organization and
repair. Together, the literature indicates that interphase microtubules
are critical regulators of nuclear structure and genome stability”
(Shokrollahi and Mekhail 2021, doi:10.1016/j.tcb.2021.03.014).
-
YY1
“There is considerable evidence that chromosome structure plays important
roles in gene control, but we have limited understanding of the proteins
that contribute to structural interactions between gene promoters and
their enhancer elements. Large DNA loops that encompass genes and their
regulatory elements depend on CTCF-CTCF interactions, but most
enhancer-promoter interactions do not employ this structural protein.
Here, we show that the ubiquitously expressed transcription factor Yin
Yang 1 (YY1) contributes to enhancer-promoter structural interactions in
a manner analogous to DNA interactions mediated by CTCF. YY1 binds to
active enhancers and promoter-proximal elements and forms dimers that
facilitate the interaction of these DNA elements. Deletion of YY1 binding
sites or depletion of YY1 protein disrupts enhancer-promoter looping and
gene expression. We propose that YY1-mediated enhancer-promoter
interactions are a general feature of mammalian gene control”
(Weintraub, Li, Zamudio et al. 2017, doi:10.1016/j.cell.2017.11.008).
-
Insulator protein CTCF (CCCTC-binding factor)
-
Structural role of RNA
One of the newest and perhaps most unexpected factors on the scene in
structuring the nucleus is RNA. “Many aspects of the underlying dynamic
nuclear architecture involve RNA as an effector molecule with structural
functions. These RNAs operate either by interacting directly or indirectly
with chromatin or participate in establishing DNA-free nuclear
subcompartments, in which certain genome-associated activities are
concentrated” (Caudron-Herger and Rippe 2012).
“Over the past few years, the contributions of RNA to organizing nuclear
structures have become clearer with the discovery that many nuclear bodies
are enriched for specific noncoding RNAs (ncRNAs); in specific cases, ncRNAs
have been shown to be essential for establishment and maintenance of these
nuclear structures. More recently, many different ncRNAs have been shown to
play critical roles in initiating the three-dimensional (3D) spatial
organization of DNA, RNA, and protein molecules in the nucleus
(Quinodoz and Guttman 2022, doi:10.1101/cshperspect.a039719).
- “RNA transcripts can act as ‘seeds’ to recruit soluble RNA-binding nuclear body components from the nucleoplasm and induce the formation of distinct types of nuclear bodies” (Caudron-Herger and Rippe 2012; Kloc, Foreman and Reddy 2011).
- “Paraspeckles play a role in regulation of gene expression via retaining certain messenger RNAs” and are built around a long noncoding RNA. Other nuclear bodies that can be formed around coding or noncoding RNAs include Cajal bodies, histone locus bodies, nuclear stress bodies, and interchromatin granules (also known as splicing speckles or SC35 domains) (Caudron-Herger and Rippe 2012).
- “Several studies point to an important role of RNA in the formation and maintenance of higher order chromatin domains that are repressive with respect to their transcriptional activity. ... This function can involve a direct structural role of the RNA itself or an indirect RNA-dependent recruitment of a chromatin-structure modifying protein”. In particular, RNAs play a role in the formation of pericentric, centromeric and telomeric domains; in the formation of polycomb group protein-based compartments; and (the most well-known example) X chromosome inactivation in female mammals (Caudron-Herger and Rippe 2012).
- However, these RNAs are not exclusively repressive: “new studies suggest that noncoding RNAs also modulate the active chromatin state. Divergent, antisense, and enhancer-like intergenic noncoding RNAs can either activate or repress gene expression by altering histone H3 lysine 4 methylation. An emerging class of enhancer-like lncRNAs may link chromosome structure to chromatin state and establish active chromatin domains” (Flynn and Chang 2012).
- And protein-coding RNAs can also play an activating role: “Long nuclear-retained coding RNAs maintain higher order chromatin in an open configuration, whereas the structure of heterochromatin domains showed a reduced dependence on RNA. The associated RNAs were found to be enriched in sequences with long 3'-UTRs, and proposed to maintain the organization of transcriptionally active chromatin compartments by stabilizing RNAP II [RNA polymerase II] transcription factories” (Caudron-Herger and Rippe 2012).
- The CTCF transcription factor helps to form chromosome loops, thereby functioning in an insulator role and in the clustering of active promoters. (See “Insulator Protein CTCF” above.) Noncoding RNAs have been found to modulate CTCF function. One RNA “is involved in a cascade of events that included epigenetic modifications and nucleosome repositioning and led to CTCF eviction”. By contrast, another RNA appears to stabilize CTCF function. And certain small (18-nucleotide) transcription initiation RNAs “were found at genomic human and mouse CTCF binding sites” (Caudron-Herger and Rippe 2012).
- “The number of RNAs with putative functions in genome organization is increasing rapidly as genome-wide mapping of nuclear RNAs is becoming a routine task” (Caudron-Herger and Rippe 2012).
- “We review findings which lead us to suggest that RNA is essentially a widespread component of interphase chromosomes. Further, RNA likely contributes to architecture and regulation, with repeat-rich ‘junk’ RNA in euchromatin promoting a more open chromatin state ... Recent findings indicate that repetitive sequences are enriched in chromosome-associated non-coding RNAs, and repeat-rich RNA shows unusual properties, including localization and stability, with similarities to XIST RNA. We suggest two frontiers in genome biology are emerging and may intersect: the broad contribution of RNA to interphase chromosomes and the distinctive properties of repeat-rich intronic or intergenic junk sequences that may play a role in chromosome structure and regulation” (Hall and Lawrence 2016, doi:10.1016/j.gde.2016.04.005).
- In sum: The structural functions of RNA “emerge as an additional regulatory layer of gene expression”. “The conformational flexibility of RNA is remarkable and makes it an ideally suited macromolecule to structure nuclear subcompartments. Single-stranded RNAs can recognize a specific DNA sequence via formation of DNA–RNA hybrids or associate to DNA duplexes in a DNA:RNA triplex. At the same time, variations in its sequence and/or secondary structure provide an essentially unlimited number of possibilities to form sites for the specific interaction with proteins. Thus, it is not surprising that RNAs can shape the nucleus in a variety of ways on different length scales ranging from single nucleotide and histone modifications to the organization of large nuclear subcompartments up to the size of a whole chromosome during X-inactivation...Accordingly, the cell type dependent expression of these RNAs is expected to play an important role for establishing specific cell functions during differentiation. Recent findings demonstrate that transcription factors involved in controlling pluripotency and differentiation regulate the expression of large intergenic ncRNAs and thus support this view” (Caudron-Herger and Rippe 2012).
“The importance of DNA shape and in particular subtle sequence-dependent
structural variations in protein-DNA interactions has only recently started to
be acknowledged in the field of genomics, connecting structural biology with
genomic research. ... Based on all these findings it is becoming increasingly
clear that in addition to the linear three-letter genetic code that is
evolutionarily constrained at the level of protein structure and function, a
structural code of DNA plays another significant role in biological function
and therefore will be subjected to evolutionary selection” (Pyle and Shakked
2011).
DNA structure does not consist of discrete, separate aspects. The
following considerations, therefore, will not always be clearly separable
from each other or, for that matter, from other factors. To illustrate the
sort of interactions to expect: “The transcriptional output of a
Hox-cofactor complex depends both on the ability of these complexes to bind
to their binding sites with high specificity, in part by reading structural
features of the DNA, and on the three-dimensional architecture of the bound
complex, which is a consequence of both protein-DNA and protein-protein
interactions” (Joshi, Sun and Mann 2010). Depending on these factors, the
“same” complex can either activate or repress a given gene.
“Variant –1 nucleosomes exhibited a preference for sequences with altered
features such as propeller twist, opening, electrostatic potential, minor
groove width, rise, stagger, helix twist, and shear and roll. Variant –1
nucleosomes that shifted downstream in KDM5B-depleted embryonic stem cells
preferred sequences with increased propeller twist, opening, electrostatic
potential, stagger, minor groove width, rise, and buckle, while –1 variant
nucleosomes that shifted upstream preferred sequences with decreased propeller
twist, opening, electrostatic potential, stagger, minor groove width, rise, and
buckle” ... Combined, these findings suggest that DNA shape predicts sequence
preferences of canonical nucleosomes and variant nucleosomes. These results
also suggest that histone DNA binding patterns such as bending or electrostatic
interactions may be influenced by posttranslational modifications such as H3K4
methylation”
(Kurup, Campeanu and Kidder 2019a, doi:10.1186/s13072-019-0266-9).
-
Supercoiling
(From an article entitled “Long-Distance Cooperative and Antagonistic RNA
Polymerase Dynamics via DNA Supercoiling”:)
“Transcription-coupled DNA supercoiling causes the following long-distance
RNAP dynamics”:
- “Multiple RNA polymerases (RNAPs) [transcribing a single gene] translocate faster than a single RNAP.”
- “Cooperation between RNAPs is not additive and occurs over long distances,” greater than two kilobases.
- “Promoter repression leads to antagonistic dynamics and premature dissociation of RNAPs.”
A study of the topological architecture of yeast genes in relation to
transcription and replication: “We found under-wound DNA at gene boundaries
and over-wound DNA within coding regions. This arrangement does not depend
on Pol II or S phase. Top2 and Hmo1 preserve negative supercoil at gene
boundaries, while Top1 acts at coding regions. Transcription generates
RNA–DNA hybrids within coding regions, independently of fork orientation.
During S phase, Hmo1 protects under-wound DNA from Top2, while Top2 confines
Pol II and Top1 at coding units, counteracting transcription leakage and
aberrant hybrids at gene boundaries. Negative supercoil at gene boundaries
prevents supercoil diffusion and nucleosome repositioning at coding regions.
DNA looping occurs at Top2 clusters. We propose that Hmo1 locks gene
boundaries in a cruciform conformation and, with Top2, modulates the
architecture of genes that retain the memory of the topological arrangements
even when transcription is repressed”
(Achar, Adhil, Choudhary et al. 2020, doi:10.1038/s41586-020-1934-4).
- Negative supercoiling of DNA, which tends to loosen the two strands of the double helix from each other, makes it easier to “unzip” the double helix and begin transcription, while positive supercoiling makes this harder. So supercoiling is a means of gene regulation. In Streptococcus pneumoniae "genes responding to changes in the level of supercoiling in a coordinated manner were found organized as functional clusters. Such an organization revealed DNA supercoiling as a general feature that controls gene expression superimposed on other kinds of more specific regulatory mechanisms” (Ferrándiz et al. 2010).
- Some gene-regulating proteins are sensitive to supercoiling and bind to sites under torsional stress (Kouzine, Sanford, Elisha-Feil et al. 2008).
- Likewise, binding of remodeling proteins is enhanced by supercoiling. (Luijsterburg et al. 2008).
- The winding of DNA around the histone cores of nucleosomes — and also the unwinding — affects the supercoiling. So also does the movement of RNA polymerase along DNA during transcription (elongation).
- “We show here that enhancer-promoter affinity and supercoiling act synergistically in increasing the fraction of time during which enhancer and promoter stay in contact. This stabilizing effect of supercoiling only acts on enhancers and promoters located in the same topological domain. We propose that the primary role of recently observed supercoiling of topological domains in interphase chromosomes of higher eukaryotes is to assure that enhancers contact almost exclusively their cognate promoters located in the same topological domain and avoid contacts with very similar promoters but located in neighbouring topological domains” (doi:10.1093/nar/gku759)
- Topoisomerases, which cut and rearrange the strands of the double helix, can alter its supercoiling, with profound implications for gene regulation. For example: “Chong et al. have now shown that, under induced conditions, transcriptional bursting — the observation that transcription occurs in random surges — is regulated through cycles of binding and dissociation of the type II topoisomerase DNA gyrase to a DNA loop in bacteria. Until now, the mechanism underlying transcriptional bursting, which has previously also been observed in eukaryotic cells, was unclear ... Chong et al. showed that positive supercoiling slows down the rate of transcription elongation and eventually stops transcription initiation. The addition of gyrase, which releases positive supercoiling, restored transcription elongation rates to the levels of controls and could reinitiate transcription. These results were further confirmed in live Escherichia coli cells. Although the mechanism in eukaryotic cells is likely to be more complicated, this study highlights the importance of chromosomal architecture on transcriptional bursting” (Lokody 2014, doi:10.1038/nrg3800).
- The enzyme, topoisomerase I, which plays a major role in managing supercoiling during transcription, tends to be inactive at promoters, and is strongly activated by phosphorylation of the C-terminal domain of the largest subunit of RNA polymerase II. Experimental results indicate that “the level of supercoiling is actively managed by the transcription machinery to melt DNA at transcription start sites, hold-back RNA polymerase II at pause sites or to accelerate elongation and help control the transcriptional output of virtually any gene” (Baranello, Wojtowicz, Cui et al. 2016, 10.1016/j.cell.2016.02.036).
-
Conformational changes in general
- “DNA has many features that allow its sequence-specific recognition by proteins. This recognition was originally thought to primarily involve specific hydrogen-bonding interactions between amino-acid side chains and bases. But it soon became clear that there was no identifiable one-to-one correspondence — that is, there was no simple code to be read. Part of the problem is that DNA can undergo conformational changes that distort the classical double helix. The resulting variations in the way that DNA bases are presented to proteins can thus affect the recognition mechanism” (Honig and Rohs 2011).
-
DNA grooves: compression and decompression
- There are small molecules that insert themselves in the minor groove and widen it, affecting access of regulatory factors. [ref ??]
- Binding of a protein regulatory factor to DNA at a given point can dramatically alter the binding of a second protein a short distance away, due to distortion of the DNA structure by the first protein. For example, “widening the [DNA major] groove by binding a protein at position zero widens the groove at +10 bp [base pairs], enhancing binding of a protein that favors a wider major groove”. By contrast, binding will be repressed at +5 bp and +15 bp. “It is now clear that the quantitative aspects of gene regulation are influenced quite substantially by the relative placement of regulatory elements. Distance changes of half a helical turn [that is, about 5 base pairs] can alter stability and [dissociation] rates by a factor of three or more. This ... makes our comparative interpretation of genome sequences even more challenging” (Crothers 2013).
- DNA stretching on the nucleosome "In vivo, stretching likely influences protein factor and small molecule recognition and modulates nucleosome positioning and chromatin compaction”. It is also sometimes associated with extreme kinking of the double helix, allowing intercalation (insertion between successive base pairs) of regulatory molecules (Tan and Davey 2011).
-
Hoogsteen base pairing
“In some DNA sequences, especially CA and TA dinucleotides, Hoogsteen
base pairs exist as transient entities that are present in thermal
equilibrium with standard Watson–Crick base pairs...the ability to flip
between Watson–Crick and Hoogsteen base pairing in free DNA is an
intrinsic property of individual sequences. This implies that some
proteins have evolved to recognize only one base-pair type, and use
intermolecular interactions to shift the equilibrium between the two
geometries” (Honig and Rohs 2011).
- “A Nature paper on spontaneous flipping of normal Watson-Crick (WC) base pairs into Hoogsteen (HG) base pairs in naked DNA under most normal conditions produced a bombshell effect throughout the world community of nucleic acids biophysicists. Just think about it: the paper claims that, on the average, roughly every hundredth base pair in DNA forms an unorthodox HG pair rather than a classical WC pair!” Yet “the claim by Nikolova et al. is based on a very solid ground” and the discovery of Hoogsteen breathing “is hugely important. The elucidation of the exact nature of the fluctuations in the DNA double helix is crucial for understanding how exactly DNA is damaged by various chemicals, [and] what is the mechanism of DNA binding with various drugs and proteins working on the level of the DNA double helix” (Frank-Kamenetskii 2011).
- Hoogsteen base pairing at the DNA binding site for the p53 “tumor suppressor” protein “generates four narrow-minor groove regions” that “are responsible for the presence of enhanced negative electrostatic potentials” which in turn are recognized by the four p53 molecules that together bind to the site (Kitayner, Rozenberg, Rohs et al. 2010).
- Article title: “Computational Mapping Reveals Dramatic Effect of Hoogsteen Breathing on Duplex DNA Reactivity with Formaldehyde” (Bohnuud, Beglov, Ngan et al. 2012).
-
Base pair opening
A less frequent and more radical “fluctuational breathing” of DNA (not to
be confused with
“DNA breathing on nucleosomes”,
discussed above) is known as “base pair opening” and plays a role in
DNA hydroxymethylation (Bohnuud,
Beglov, Ngan et al. 2012; Frank-Kamenetskii 2011).
- Bendability of double helix
-
Transient DNA strand separation (breathing)
“Growing evidence suggests that, in the cell, the double-stranded DNA
(dsDNA) is subject to local transient strand separations (breathing) that
contribute to chromatin functions”. “Our extensive experimental and
computational efforts point to sequence-specific DNA breathing dynamics,
that is composed of local transient openings of the double helix (aka DNA
bubbles), as an essential factor for transcriptional activity in mammalian
promoters” (Alexandrov, Fukuyo, Lange et al. 2012). Strand separation is
often referred to as “melting” of the DNA.
- Various features of strand separation, or DNA breathing, appear to have a direct influence on binding of transcription factors to DNA. Regarding the ubiquitous transcription factor YY1 in human cells: “We find that YY1 binding in cells depends not only on the availability of direct points-of-contact with DNA, as previously reported, but also on a particular DNA breathing profile, that depends on the flanking sequence and can be altered by single SNPs [single-nucleotide polymorphisms — changes in a single ‘letter’ of DNA] outside of the direct recognition sequence”. Genomic flanking sequence variations and SNPs “may exert long-range effects on DNA dynamics and predetermine YY1 binding. ... YY1 has a fundamental role in essential biological processes by activating, initiating or repressing transcription depending upon the sequence context it binds” (Alexandrov, Fukuyo, Lange et al. 2012).
-
DNA structural variants
“Structural variants (SVs) are an important source of human genome diversity
... We estimate that common SVs are causal at 2.66% of eQTLs [expression
quantitative trait loci], a 10.5-fold enrichment relative to their abundance
in the genome. Duplications and deletions were the most impactful variant
types, whereas the contribution of mobile element insertions was small
(0.12% of eQTLs, 1.9-fold enriched). Multitissue analysis of eQTLs revealed
that gene-altering SVs show more constitutive effects than other variant
types, with 62.09% of coding SV-eQTLs active in all tissues with eQTL
activity compared with 23.08% of coding SNV- [single-nucleotide variant] and
indel-eQTLs. Noncoding SVs, SNVs and indels show broadly similar patterns.
We also identified 539 rare SVs associated with nearby gene expression
outliers. Of these, 62.34% are noncoding SVs that affect gene expression but
have modest enrichment at regulatory elements, showing that rare noncoding
SVs are a major source of gene expression differences but remain difficult
to predict from current annotations. Both common and rare SVs often affect
the expression of multiple genes: SV-eQTLs affect an average of 1.82 nearby
genes, whereas SNV- and indel-eQTLs affect an average of 1.09 genes, and
21.34% of rare expression-altering SVs show effects on two to nine different
genes. We also observe significant effects on rare gene expression changes
extending 1 Mb from the SV. This provides a mechanism by which individual
SVs may have strong or pleiotropic effects on phenotypic variation” (Scott,
Chiang and Hall 2021, doi:10.1101/gr.275488.121).
“Structural variants (SVs) can affect protein-coding sequences as well as
gene regulatory elements. However, SVs disrupting protein-coding sequences
that also function as cis-regulatory elements remain largely
uncharacterized. Here, we show that craniosynostosis patients with SVs
containing the histone deacetylase 9 (HDAC9) protein-coding sequence are
associated with disruption of TWIST1 regulatory elements that reside within
the HDAC9 sequence ... Deletions of either Twist1 enhancers or CTCF site
within the Hdac9 protein-coding sequence led to decreased Twist1 expression
and altered anterior/posterior limb expression patterns of SHH [sonic
hedgehog] pathway genes. This decreased Twist1 expression results in a
smaller sized and asymmetric skull and polydactyly that resembles Twist1+/−
mouse phenotype. Chromatin conformation analysis revealed that the Twist1
promoter interacts with Hdac9 sequences that encompass Twist1 enhancers and
a CTCF site, and that interactions depended on the presence of both
regulatory regions. Finally, a large inversion of the entire Hdac9 sequence
in mice that does not disrupt Hdac9 expression but repositions Twist1
regulatory elements showed decreased Twist1 expression and led to a
craniosynostosis-like phenotype and polydactyly. Thus, our study ...
suggests that SVs encompassing protein-coding sequences could lead to a
phenotype that is not attributed to its protein function but rather to a
disruption of the transcriptional regulation of a nearby gene”
(Hirsch, Dahan, D'haene et al. 2022, doi:10.1101/gr.276196.121).
-
Electrical structure of DNA
“Chromatin is a negatively charged polymer that produces electrostatic
repulsion between adjacent regions because only about half of the DNA
negative charges derived from the phosphate backbone are neutralized by the
basic core histones. For further folding of chromatin, the remaining
negative charges have to be neutralized by other factors, such as linker
histones, cations, and other positively charged molecules. Therefore, the
chromatin structure can be dynamically changed depending on the
electrostatic state of its environment. Note that this change in chromatin
structure is critical for gene expression because it directly governs access
to the DNA and therefore impacts how the DNA is scanned and read.
“Recent growing evidence has shifted our view of chromatin from one in which it has a static crystal-like structure to one in which it occupies a more dynamic liquid-like state” (Maeshima, Ide, Hibino and Sasai 2016, doi:10.1016/j.gde.2015.11.006).
- There are regulatory proteins that recognize the local electrical “signatures” of particular stretches of DNA. [particulars ??]
-
DNA-RNA duplexes (triplexes)
- See also “Promoter-associated RNAs” under NONCODING RNA above. The authors of the study cited there write, "A triplex-mediated mechanism of DNA methylation and gene silencing is particularly intriguing considering that the vast majority of the transcriptional output of the human genome represents noncoding RNA. Therefore, ncRNAs can provide unique sequence specificity via triplex formation and may function as binding platforms for chromatin-modifying enzymes without the need of adaptor proteins”.
- Fragile X syndrome is considered the most common genetic form of mental retardation. A CGG trinucleotide-repeat expansion adjacent to the FMR1 gene promoter results in epigenetic silencing of the gene. What happens is that the expanded repeat is included in the 5'-untranslated region of the FMR1 mRNA, which then hybridizes to the complementary CGG-repeat portion of the gene, forming an RNA-DNA duplex. A CGG-related hairpin structure in the mRNA must be unfolded and linearized in order for the hybridization and gene silencing to occur. All this is observed to happen in human embryonic stem cells upon their differentiation into neuronal cells (Colak, Zaninovic, Cohen et al. 2014).
- “RNA can directly bind to purine-rich DNA via Hoogsteen base pairing, forming a DNA:RNA triple helical structure that anchors the RNA to specific sequences and allows guiding of transcription regulators to distinct genomic loci. ... High-throughput sequencing and computational analysis of DNA-associated RNA revealed a large set of RNAs which originate from non-coding and coding loci, including super-enhancers and repeat elements. Combined analysis of DNA-associated RNA and RNA-associated DNA identified genomic DNA:RNA triplex structures. The results suggest that triplex formation is a general mechanism of RNA-mediated target-site recognition, which has major impact on biological functions” (Cetin, Kuo, Ribarska et al. 2019, doi:10.1093/nar/gky1305).
-
Double-stranded RNAs
- “We show that hundreds of putative natural double-stranded RNAs (ndsRNAs) are expressed from interspersed genomic locations and respond to cellular cues. We demonstrate that a subset of ndsRNAs localize in the nucleus and, in their double-stranded form, interact with nuclear proteins. Detailed characterization of an ndsRNA (nds-2a) revealed that this molecule displays differential localization throughout the cell cycle and directly interacts with RCC1 and RAN and, through the latter, with the mitotic RANGAP1–SUMO1–RANBP2 complex. Notably, altering nds-2a levels led to postmitotic abnormalities, mitotic catastrophe and cell death, thus supporting a mitosis-related role. Altogether, our study reveals a hitherto-unrecognized class of RNAs that potentially participate in major biological processes in human cells” (Portal, Pavet, Erb and Gronemeyer 2015, doi:10.1038/nsmb.2934).
- On double-stranded RNA-binding proteins (dsRBPs): “Our results demonstrate that despite the highly conserved dsRNA binding domains, the dsRBPs exhibit diverse substrate specificities and dynamic properties when in contact with different RNA substrates. While TRBP and ADAR1 have a preference for binding simple duplex RNA, ADAD2 and Staufen1 display higher affinity to highly structured RNA substrates. Upon interaction with RNA substrates, TRBP and Staufen1 exhibit dynamic sliding whereas two deaminases ADAR1 and ADAD2 mostly remain immobile when bound ... Collectively, our study highlights the diverse nature of substrate specificity and mobility exhibited by dsRBPs that may be critical for their cellular function” (Wang, Vukovic, Koh et al. 2015, doi:10.1093/nar/gkv726).
-
DNA/RNA R-loops
“Remarkably RNA can ... anneal to its genomic template co- or
post-transcriptionally to generate an RNA–DNA hybrid and a displaced
single-stranded DNA. These unusual nucleic acid structures are called
R-loops. Studies in the last decades concentrated on the detrimental effects
of R-loop formation, particularly on genome stability. In fact, R-loops are
thought to play a role in several human diseases like cancer and
neurodegenerative syndromes. But recent data has revealed that R-loops can
also have a positive impact on cell processes, like regulating gene
expression, chromosome structure and DNA repair” (Costantino and Koshland
2015, doi:10.1016/j.ceb.2015.04.008).
“RNA–DNA hybrids are generated during transcription, DNA replication and DNA
repair and are crucial intermediates in these processes. When RNA–DNA
hybrids are stably formed in double-stranded DNA, they displace one of the
DNA strands and give rise to a three-stranded structure called an R-loop.
R-loops are widespread in the genome and are enriched at active genes.
R-loops have important roles in regulating gene expression and chromatin
structure, but they also pose a threat to genomic stability, especially
during DNA replication. To keep the genome stable, cells have evolved a slew
of mechanisms to prevent aberrant R-loop accumulation. Although R-loops can
cause DNA damage, they are also induced by DNA damage and act as key
intermediates in DNA repair such as in transcription-coupled repair and
RNA-templated DNA break repair. When the regulation of R-loops goes awry,
pathological R-loops accumulate, which contributes to diseases such as
neurodegeneration and cancer”
(Petermann, Lan and Zou 2022, doi:10.1038/s41580-022-00474-x).
“R-loop misregulation is associated with DNA damage, transcription
elongation defects, hyper-recombination and genome instability. In contrast
to such ‘unscheduled’ R-loops, evidence is mounting that cells harness the
presence of RNA–DNA hybrids in scheduled, ‘regulatory’ R-loops to promote
DNA transactions, including transcription termination and other steps of
gene regulation, telomere stability and DNA repair. R-loops formed by
cellular RNAs can regulate histone post-translational modification and may
be recognized by dedicated reader proteins. The two-faced nature of R-loops
implies that their formation, location and timely removal must be tightly
regulated”
(Niehrs and Luke 2020, doi:10.1038/s41580-019-0206-3).
- “R-loops, which have been considered to be rare and potentially harmful transcriptional by-products, are now shown to be needed for antisense transcription and to induce repressive chromatin marks that reinforce pausing of transcription and thereby enhance its termination” (blurb in Nature for doi:10.1038/nature13787). The authors write, “We predict that R-loops promote a chromatin architecture that defines the termination region for a substantial subset of mammalian genes.”
- “We show that transcription-blocking DNA lesions promote chromatin displacement of late-stage spliceosomes and initiate a positive feedback loop centred on the signalling kinase ATM. We propose that initial spliceosome displacement and subsequent R-loop formation is triggered by pausing of RNA polymerase at DNA lesions. In turn, R-loops activate ATM, which signals to impede spliceosome organization further and augment ultraviolet-irradiation-triggered alternative splicing at the genome-wide level” (Tresini1, Warmerdam, Kolovos et al. 2015, doi:10.1038/nature14512).
- “Chen et al. studied the chromatin-activating complex Tip60–p400 (an acetyltransferase) and found that it was able to bind to nascent transcripts near their initiation start sites, and that this binding was enhanced by the presence of DNA:RNA hybrids (R loops) between the transcript and the DNA template. At the same time, binding of the Polycomb repressive complex 2 (PRC2) was found to be inhibited by the formation of R loops. Thus, the presence of R loops near the 5′ ends of transcribed genes affects the recruitment of chromatin remodellers to these sites, thereby shaping chromatin structure and influencing transcription. Moreover, the authors provided evidence that the disruption of R loops perturbed stem cell differentiation, indicating that the absence of R loops can lead to global changes in gene expression” (Strzyz 2015, doi:10.1038/nrm4094).
- “Epigenomic profiling revealed that R loops associate with specific epigenomic signatures: at promoters, R loops associate with an open, histone H3 lysine 4 (H3K4) hypermethylated and hyperacetylated state characteristic of strong CpG island promoters; at terminators, R loops associate with an enhancer- and insulator-like state; and R-loop formation seems to be a conserved hallmark of a broad class of transcription terminators” (Koch 2016, doi:10.1038/nrg.2016.92).
- “R‐loops, formed by co‐transcriptional DNA–RNA hybrids and a displaced DNA single strand (ssDNA), fulfill certain positive regulatory roles but are also a source of genomic instability. One key cellular mechanism to prevent R‐loop accumulation centers on the conserved THO/TREX complex, an RNA‐binding factor involved in transcription elongation and RNA export that contributes to messenger ribonucleoprotein (mRNP) assembly, but whose precise function is still unclear. To understand how THO restrains harmful R‐loops, we searched for new THO‐interacting factors. We found that human THO interacts with the Sin3A histone deacetylase complex to suppress co‐transcriptional R‐loops, DNA damage, and replication impairment. Functional analyses show that histone hypo‐acetylation prevents accumulation of harmful R‐loops and RNA‐mediated genomic instability” (Salas‐Armenteros, Pérez‐Calero, Bayona‐Feliu et al. 2017, doi:10.15252/embj.201797208).
- “m6A RNA modification as a new player in R-loop regulation”: “RNA:DNA hybrids that form across genomes control a wide range of biological processes. A new study shows that N6-methyladenosine (m6A) modification on the RNA moieties regulates the formation and genome integrity of these hybrids. This finding opens a new avenue of research on how RNA modifications (the ‘epitranscriptome’) can help control genome maintenance” (TOC blurb for Marnef and Legube 2020, doi:10.1038/s41588-019-0563-z).
- “Enhancers generate bidirectional noncoding enhancer RNAs (eRNAs) that may regulate gene expression ... Here, we report a 5′ capped antisense eRNA PEARL (Pcdh eRNA associated with R-loop formation) that is transcribed from the protocadherin (Pcdh) α HS5-1 enhancer region ... we found that PEARL regulates Pcdhα gene expression by forming local RNA–DNA duplexes (R-loops) in situ within the HS5-1 enhancer region to promote long-distance chromatin interactions between distal enhancers and target promoters. In particular, increased levels of eRNA PEARL via perturbing transcription elongation factor SPT6 lead to strengthened local three-dimensional chromatin organization within the Pcdh superTAD” (Zhou, Xu, Zhang and Wu 2021, doi:10.1101/gad.348621.121 ).
- “R-loops, formed transiently during gene transcription, are tightly controlled to avoid conflict with ongoing processes. Marchena-Cruz et al. identified DExD/H box RNA helicase DDX47 using a new R-loop resolving screen and defined a unique role for this helicase in nucleolar R-loops and its interplay with senataxin (SETX) and DDX39B” (Yu and Richard 2023, doi:10.1016/j.tcb.2023.03.001).
-
DNA G-quadruplexes
G-quadruplexes comprise a diverse group of non-canonical DNA
structures that have in common a set of guanine tetrads (G4-DNA). Guanine
(represented by the letter G) normally pairs with cytosine (C) in the
canonical DNA double helix. That is, in a common analogy: a G-C pair forms
one type of “rung” in the the spiraling “ladder” of the double helix, with
the side-rails of the ladder constituting the DNA “backbone”. By contrast,
given the presence of sufficient guanines in a length of DNA, a more
complex structure can form where the double helix bends back on itself and
a series of stacked guanine tetrads is formed between four “side-rails”.
Many variations are possible, depending on the directions of the various
“side-rails” (parallel or anti-parallel) and also depending on whether the
structure is formed from a single double helix bending back on itself or
multiple DNA molecules joining together.
Regarding our developing understanding of G-quadruplexes (G4s) as epigenetic
factors: “First, persistent G4s (e.g., those stabilized by exogenous
ligands) were linked to the loss of the histone code. More recently,
transient G4s (i.e., those formed upon replication or transcription and
unfolded rapidly by helicases) were implicated in CpG island methylation
maintenance and de novo CpG methylation control. The most recent data
indicate that there are direct interactions between G4s and chromatin
remodeling factors. Finally, multiple findings support the indirect
participation of G4s in chromatin reshaping via interactions with
remodeling‐related transcription factors (TFs) or damage responders”
(Varizhuk, Isaakova and Pozmogova 2019, doi:10.1002/bies.201900091).
“We find that native G4 [G-quadruplex] signals are cell type–specific and
are associated with transcriptional regulatory elements carrying active
epigenetic modifications. Drug-induced promoter-proximal RNA polymerase II
pausing promotes nearby G4 formation. In contrast, G4 stabilization by
G4-targeted ligands globally reduces RNA polymerase II occupancy at gene
promoters as well as nascent RNA synthesis. Moreover, ligand-induced G4
stabilization modulates chromatin states and impedes transcription
initiation via inhibition of general transcription factors loading to
promoters. Together, our study reveals a reciprocal genome-wide regulation
between native G4 dynamics and gene transcription”
(Li, Wang, Yin et al. 2021, doi:10.1101/gr.275431.121 ).
- In studies of different organisms, G-quadruplexes showed “exquisite specificity for heterochromatin”. “The unexpected presence of G4 structures in heterochromatin and the difference in G4 staining between somatic cells and stem cells with germline DNA in ciliates, flatworms, flies and mammals point to a conserved role for G4 structures in nuclear organization and cellular differentiation” (Hoffmann, Moshkin, Mouton et al. 2016, doi:10.1093/nar/gkv900).
- “A genome-wide survey of the evolutionary conservation of DNA motifs indicated that G4 [G-quadruplex] DNA motifs are significantly conserved. Increasing evidence suggests an important role of G4 DNA structures in regulating gene expression. Interestingly, G4 DNA structures are more enriched in promoters than other regions of genomic DNA, especially in genes involved in development, survival and proliferation” (Huang, Smaldino, Zhang et al. 2012).
- Removal of G-quadruplexes from the promoter of a tumorigenesis gene or from the 5’-UTR of its mRNA transcript increases expression of the gene and facilitates tumor formation (Huang, Smaldino, Zhang et al. 2012).
- Extensive and multifaceted studies have shown that “non-canonical guanine quadruplex structures are not only predominant but also conserved among bacterial and mammalian promoters. ... The quadruplex motif, and not the disrupted-motif, enhanced transcription in human cell lines of different origin. Together, these findings build direct support for quadruplex-mediated transcription and suggest quadruplex-SNPs may play significant role in mechanistically understanding variations in gene expression among individuals”. Disruption of the quadruplexes resulted in inhibition of transcription. Evidence “suggests the possibility that subtle changes in the G-quadruplex form/stability leads to relatively pronounced gene expression changes due to altered DNA binding of transcription factors” (Baral, Kumar, Halder et al. 2012).
- “We show that G-quadruplex formation can be remotely induced by downstream transcription events that are thousands of base pairs away. The induced G-quadruplexes alter protein recognition and cause transcription termination at the local region. These results suggest that a G-quadruplex-forming sequence can serve as a sensor or receiver to sense remote DNA tracking activity in response to the propagation of mechanical torsion in a DNA double helix. We propose that the G-quadruplex formation may provide a mean for long-range sensing and communication between distal genomic locations to coordinate regulatory transactions in genomic DNA” (Zhang, Liu, Zheng et al. 2013).
- “Applying [a new technique called] G4-seq in primary lymphocytes revealed >500,000 G4 structures in the human genome. Notably, ~70% of these were not predicted computationally, because they contain bulges or long loops. Such non-canonical G4 structures were exceptionally prevalent in gene regulatory regions, especially in 5' UTRs and splicing sites, and also in oncogenes and (other) regions that are amplified in cancers” (Zlotorynski 2015, doi:10.1038/nrm4052).
- “Cancer stem cells (CSCs) have been identified in several solid malignancies and are now emerging as a plausible target for drug discovery ... the expression of [the gene] CD133 was reported to be responsible for conferring CSC aggressiveness. Here, we identified two G-rich sequences localized within the introns 3 and 7 of the CD133 gene able to form G-quadruplex (G4) structures, bound and stabilized by small molecules. We further showed that treatment of patient-derived colon CSCs with G4-interacting agents triggers alternative splicing that dramatically impairs the expression of CD133. Interestingly, this is strongly associated with a loss of CSC properties, including self-renewing, motility, tumor initiation and metastases dissemination ... In conclusion, we provided the first proof of the existence of G4 structures within the CD133 gene that can be pharmacologically targeted to impair CSC aggressiveness” (Zizza, Cingolani, Artuso et al. 2016, doi:10.1093/nar/gkv1122).
-
More on alternative DNA structures
“Repetitive elements in the human genome, once considered ‘junk DNA’, are
now known to adopt more than a dozen alternative (that is, non-B) DNA
structures, such as self-annealed hairpins, left-handed Z-DNA,
three-stranded triplexes (H-DNA) or four-stranded guanine quadruplex
structures (G4 DNA). These dynamic conformations can act as functional
genomic elements involved in DNA replication and transcription, chromatin
organization and genome stability. In addition, recent studies have revealed
a role for these alternative structures in triggering error-generating DNA
repair processes, thereby actively enabling genome plasticity. As a driving
force for genetic variation, non-B DNA structures thus contribute to both
disease aetiology and evolution”
(Wang and Vasquez 2023, doi:10.1038/s41576-022-00539-9).
-
DNA damage repair
“The idea that signal-dependent transcription might involve the generation
of transient DNA nicks or even breaks in the regulatory regions of genes,
accompanied by activation of DNA damage repair pathways, would seem to be
counterintuitive, as DNA damage is usually considered harmful to cellular
integrity. However, recent studies have generated a substantial body of
evidence that now argues that programmed DNA single- or double-strand breaks
can, at least in specific cases, have a role in transcription regulation”.
(Puc, Aggarwal and Rosenfeld 2017, doi:10.1038/nrm.2017.43).
According to the authors, relief of DNA torsional stress plays a role in
promoter and enhancer activation.
“It is becoming increasingly clear that many DNA repair factors function
directly in transcription regulation. In particular, ‘programmed’ DNA nicks
and breaks seem to have a strategic role in the regulation of gene
expression. DNA breaks appear to be employed not only in hormone-induced
transcription, but also in the regulation of some tissue-specific genes.
Although the DNA topoisomerases TOP1 and TOP2 appear to be the most widely
used DNA cleaving enzymes, other DNA cleaving enzymes can also be used.
Together, programmed DNA breaks promote not only transcription elongation by
Pol II but probably also the assembly of large, multiprotein regulatory
complexes and the formation of long-range interactions between transcription
units”
(Puc, Aggarwal and Rosenfeld 2017, doi:10.1038/nrm.2017.43).
“The organization of the genome into topologically associated domains
suggests new possible roles for DNA breaks, which include, for example, the
untangling of CCCTC-binding factor- and cohesin-mediated chromatin loops,
and mediating interactions between promoters and enhancers on different
loops”
(Puc, Aggarwal and Rosenfeld 2017, doi:10.1038/nrm.2017.43).
The following suggest just how complex and crucial is the conjunction of
gene transcription and DNA damage repair:
“Transcription-coupled DNA repair removes bulky DNA lesions from the
genome, and protects cells against ultraviolet (UV) irradiation.
Transcription-coupled DNA repair begins when RNA polymerase II (Pol II)
stalls at a DNA lesion and recruits the Cockayne syndrome protein CSB, the
E3 ubiquitin ligase, CRL4CSA and UV-stimulated scaffold protein A (UVSSA).
... Stalling of Pol II at a DNA lesion triggers replacement of the
elongation factor DSIF by CSB, which binds to PAF and moves upstream DNA to
SPT6. The resulting elongation complex, ECTCR, uses the CSA-stimulated
translocase activity of CSB to pull on upstream DNA and push Pol II forward.
If the lesion cannot be bypassed, CRL4CSA spans over the Pol II clamp and
ubiquitylates the RPB1 residue K1268, enabling recruitment of TFIIH to UVSSA
and DNA repair. Conformational changes in CRL4CSA lead to ubiquitylation of
CSB and to release of transcription-coupled DNA repair factors before
transcription may continue over repaired DNA”
(Kokic, Wagner, Chernev et al. 2021, doi:10.1038/s41586-021-03906-4).
-
RNA structure and dynamics
“Single-stranded RNA molecules fold into extraordinarily complicated
secondary and tertiary structures as a result of intramolecular base
pairing. In vivo, these RNA structures are not static. Instead, they are
remodeled in response to changes in the prevailing physicochemical
environment of the cell and as a result of intermolecular base pairing and
interactions with RNA-binding proteins ... Analyses of RNA structuromes in
HIV, yeast, Arabidopsis, and mammalian cells and tissues have
revealed regulatory effects of RNA structure on messenger RNA (mRNA)
polyadenylation, splicing, translation, and turnover. Application of new
methods for genome-wide identification of mRNA modifications, particularly
methylation and pseudouridylation, has shown that the RNA ‘epitranscriptome’
both influences and is influenced by RNA structure”
(Bevilacqua, Ritchey, Su and Assmann 2016,
doi:10.1146/annurev-genet-120215-035034).
“Base-pairing changes play a simple but powerful role in gene regulation in
part by sequestering or exposing nucleotides that can interact with other
RNAs, proteins, or other cellular constituents. RNA structure thus can be
considered as another, previously hidden, layer of the genetic code that we
are only just beginning to understand”
(Bevilacqua, Ritchey, Su and Assmann 2016,
doi:10.1146/annurev-genet-120215-035034).
Some RNA-binding proteins target specific RNA sequences, “but even in these
cases, the folding structure of the RNA ... has a major bearing on whether
the regulatory proteins can bind to the targets (Li, Quon, Lipshitz and
Morris 2010).
“Changes to the conformation of coding and non-coding RNAs form the basis
of elements of genetic regulation and provide an important source of
complexity, which drives many of the fundamental processes of life. ... The
conventional view that one sequence codes for one structure and one
function is being replaced by a dynamic view of RNA as a pre-existing
superposition of conformational states that can be resolved into a directed
and synchronized motion by dedicated cellular machinery, leading to a broad
range of functional outcomes”. And again: “Early X-ray structures of RNA
contained indications of the importance of conformational dynamics: large
changes in the helical arms of transfer RNA were observed on the binding of
tRNA synthetase, and changes in the conformation of ribozymes needed to be
invoked to envision catalytically active states. However, no one could have
anticipated the existence of new genetic circuits that are based on RNA
conformational switches, or that the ‘acrobatic’ nature of a biopolymer
that consists of only four chemically similar nucleotides would be at the
centre of a complex macromolecular structure such as the ribosome”
(Dethoff, Chugh, Mustoe and Al-Hashimi 2012).
“Although the basic principles of gene expression were established some 60
years ago, recent research has revealed a surprising complexity in the
control of gene activity. Many of these gene regulatory mechanisms occur at
the level of the mRNA, including sophisticated gene control tasks mediated
by structured mRNA elements. We now know that mRNA folds can serve as
highly specific receptors for various types of molecules, as exemplified by
metabolite-binding riboswitches, and interfere with pro- and eukaryotic
gene expression at the level of transcription, translation, and RNA
processing. Gene regulation by structured mRNA elements comprises versatile
strategies including self-cleaving ribozymes, RNA-folding-mediated
occlusion or presentation of cis-regulatory sequences, and sequestration of
trans-acting factors including other RNAs and proteins” (Wachter 2014).
The importance of secondary structures in mRNAs is evidenced by the
evolutionary conservation of such structures — that is, mutations altering
the structures tend to be weeded out over evolutionary time. (Chursov,
Frishman and Shneider 2013, in a study of E. coli).
“We investigate the possibility that mRNA structures facilitate the 3'-end
processing of thousands of human mRNAs by juxtaposing poly(A) signals (PASs)
and cleavage sites that are otherwise too far apart. We find that RNA
structures are predicted to be more prevalent within these extended 3'-end
regions than within PAS-upstream regions and indeed are substantially more
folded within cells, as determined by intracellular probing. Analyses of
thousands of ectopically expressed variants demonstrate that this folding
both enhances processing and increases mRNA metabolic stability ...
Structure-controlled processing can also regulate neighboring gene
expression. Thus, RNA structure has widespread roles in mammalian mRNA
biogenesis and metabolism”
(Wu and Bartel 2017, doi:10.1016/j.cell.2017.04.036).
“RNA molecules fold into complex three-dimensional structures that sample
alternate conformations ranging from minor differences in tertiary structure
dynamics to major differences in secondary structure. This allows them to
form entirely different substructures with each population potentially
giving rise to a distinct biological outcome. The substructures can be
partitioned along an existing energy landscape given a particular static
cellular cue or can be shifted in response to dynamic cues such as ligand
binding. We review a few key examples of RNA molecules that sample alternate
conformations and how these are capitalized on for control of critical
regulatory functions” (Wu and D’Souza 2020, doi:10.1101/cshperspect.a032425).
- “RNA structure can regulate splicing in at least three ways: (a) by affecting assembly of the actual spliceosome; (b) as a component of motifs recognized by splicing factors, i.e., proteins that regulate splicing; and (c) by altering base pairing in ways that affect splicing choice or efficiency, independent of protein binding” (Bevilacqua, Ritchey, Su and Assmann 2016, doi:10.1146/annurev-genet-120215-035034).
- “Here, we consider how the coupling of RNA modifications and structures shapes RNA-protein interactions at different steps of the gene expression process” (Lewis, Pan and Kalsotra 2017, doi:10.1038/nrm.2016.163).
-
Stem loops and other secondary structures
- Using new techniques to investigate RNA structures bound by Staufen 1 in human cells, researchers have uncovered “a dominance of intra-molecular RNA duplexes, a depletion of duplexes from coding regions of highly translated mRNAs, an unexpected prevalence of long-range duplexes in 3′ untranslated regions (UTRs), and a decreased incidence of single nucleotide polymorphisms in duplex-forming regions. We also discover a duplex spanning 858 nucleotides in the 3′ UTR of the X-box binding protein 1 (XBP1) mRNA that regulates its cytoplasmic splicing and stability. Our study reveals the fundamental role of mRNA secondary structures in gene expression” (Sugimoto, Vigilante, Darbo et al. 2015, doi:10.1038/nature14280).
- In one study, “[RNA] sequences containing binding motifs for RBPs [RNA-binding proteins] underwent structural rearrangements in vivo, suggesting that RNA structure or accessibility was being modulated by RBP binding” (Burgess 2015, doi:10.1038/nrg3939 — reporting on work by Spitale, Flynn, Zhang et al. 2015, doi:10.1038/nature14263).
- “Double stem-loop and other structural motifs [in long and short noncoding RNA] recruit Polycomb complex for gene silencing” in mammals (Wan, Kertesz, Spitale et al. 2011).
- Binding of proteins to stem-loop structures in mRNA regulates alternative splicing in mammals (Wan, Kertesz, Spitale et al. 2011).
- Also in mammals, RNA structure contributes to the localization of the RNA to various organelles or regions of the cell (which, of course, helps to determine the function of the RNA) (Wan, Kertesz, Spitale et al. 2011).
- A stem-loop in the 3'-UTR [untranslated region] of an RNA can prevent RNA decay in mammals (Wan, Kertesz, Spitale et al. 2011).
- “Half-lives of mRNA isoforms from the same gene, including nearly identical isoforms, often vary widely. Based on clusters of isoforms with different half-lives, we identify hundreds of sequences conferring stabilization or destabilization upon mRNAs terminating downstream. One class of stabilizing element is a polyU sequence that can interact with poly(A) tails, inhibit the association of poly(A)-binding protein, and confer increased stability upon introduction into ectopic transcripts. More generally, destabilizing and stabilizing elements are linked to the propensity of the poly(A) tail to engage in doublestranded structures. Isoforms engineered to fold into 3' stem-loop structures not involving the poly(A) tail exhibit even longer half-lives. We suggest that double-stranded structures at 3' ends are a major determinant of mRNA stability” (Geisberg, Moqtaderi, Fan et al. 2014).
- Secondary structure in the 5-UTR of a target mRNA is often necessary for miRNA translational repression and mRNA degradation (Meijer, H. A., Kong, Lu et al. 2013).
- See also “RNA structure” under DECISION-MAKING RELATING TO TRANSLATION . above.
- “LincRNA-p21 is a long intergenic non-coding RNA (lincRNA) involved in the p53-mediated stress response. We sequenced the human lincRNA-p21 (hLincRNA-p21) and found that it has a single exon that includes inverted repeat Alu elements (IRAlus). Sense and antisense Alu elements fold independently of one another into a secondary structure that is conserved in lincRNA-p21 among primates. Moreover, the structures formed by IRAlus are involved in the localization of hLincRNA-p21 in the nucleus, where hLincRNA-p21 colocalizes with paraspeckles. Our results underscore the importance of IRAlus structures for the function of hLincRNA-p21 during the stress response” (Chillón and Pyle 2016, doi:10.1093/nar/gkw599).
- “mRNAs containing exceptionally stable secondary structure elements typically encode compact proteins. [Experimental results indicate] an important role of mRNA secondary structure elements in the control of protein folding ... These findings suggest a model in which the mRNA structure, particularly exceptionally stable RNA structural elements, act as gauges of protein co-translational folding by reducing ribosome speed when the nascent peptide needs time to form and optimize the core structure” (Faure, Ogurtsov, Shabalina and Koonin 2016, doi:10.1093/nar/gkw671).
-
RNA G-quadruplexes
G-quadruplexes — tetrahelix structures rich in guanine — can form in
the 5'-untranslated region (5'-UTR) of mRNA molecules. Their formation
can, by various routes repress translation of the mRNAs. And now it’s
been found that “G4 [G-quadruplex] structures are abundant within
3'-UTRs” and they make “diverse contributions to mRNA processing”
(Beaudoin and Perreault 2013).
“Recent advances have demonstrated that RNA G-quadruplexes are key
players in various cellular functions, including telomere homeostasis,
pre-mRNA processing (splicing and polyadenylation), mRNA targeting, RNA
turnover and translation” (Yu, Teulade-Fichou and Olsthoorn 2014).
“A direct correlation between thermodynamic stability of RNA G4s in
5'-UTRs and their ability to repress translation has been shown,
suggesting that RNA G-quadruplexes can act as tunable roadblocks to
control gene expression by affecting ribosome scanning” (Yu,
Teulade-Fichou and Olsthoorn 2014).
“Recent advances have highlighted new functions of RG4s [RNA
G-quadruplexes] in the regulation of RNA expression in mitochondria, in
phase separation mechanisms underscoring the formation of membrane-less
organelles, and in chemical modifications within transcripts resulting in
dynamic shaping of post-transcriptional gene expression pathways.
RG4-binding proteins are key players in regulating the dynamic
equilibrium of their formation/dissolution in the cell, controlling their
biological functions and driving their deregulation associated with human
diseases.” “There is recent evidence that these structures, in synergy
with RBPs [RNA-binding proteins], can be dynamically regulated in
cellulo and play a key role in regulating chemical RNA composition or
cell compartmentalization (both membrane-bound and membrane-less)”
(Dumas, Herviou, Dassi et al. 2021, doi:10.1016/j.tibs.2020.11.001).
“Guanine-rich regions of DNA or RNA can form structures with two or more
consecutive G-quartets called G-quadruplexes (GQ). Recent studies reveal
the potential for these structures to aggregate in vitro. Here, we report
effects of in vivo concentrations of additives — amino acids,
nucleotides, and crowding agents — on the structure and solution behavior
of RNAs containing GQ-forming sequences. We found that cytosine
nucleotides destabilize a model GQ structure at biological salt
concentrations, while free amino acids and other nucleotides do not do so
to a substantial degree. We also report that the tendency of folded GQs
to form droplets or to aggregate depends on the nature of flanking
sequence and the presence of additives. Notably, in the presence of
biological amounts of polyamines, flanking regions on the 5′-end of the
RNA drive more droplet-like phase separation, while flanking regions on
the 3′-end, as well as both the 5′- and 3′-ends, induce more condensed,
granular structures. Finally, we provide an example of a biological
sequence in the presence of polyamines and show that crowders such as PEG
and dextran can selectively cause its phase separation”
(Williams, Dickson, Lagoa-Miguel and Bevilacqua 2022,
doi:10.1261/rna.079196.122).
- “Clearly the 5'-UTR G-quadruplexes represent a class of translational repressors that is broadly distributed in the cell” (Beaudoin and Perreault 2010).
- G-quadruplex structures in mammalian RNAs, as well as the binding of proteins to various structural features, shape gene expression by affecting the translation of the RNAs (Wan, Kertesz, Spitale et al. 2011).
- “G-quadruplex structures cause transcription termination” of mammalian mitochondrial RNA (Wan, Kertesz, Spitale et al. 2011).
- While most studies have focused on inhibition of translation due to G-quadruplexes, “there are cases where RNA G-quadruplex formation has been shown to actually promote translation” (Bugaut and Balasubramanian 2012).
- As is true of regulatory factors in general, G-quadruplexes themselves can be regulated: “Recent publications have provided proof-of-concept for the inhibition of translation by small molecules that target G-quadruplexes in the 5'-UTR of RNA transcripts” (Bugaut and Balasubramanian 2012).
- Proteins have also been found that preferentially bind to G-quadruplexes and presumably perform a regulatory role (Bugaut and Balasubramanian 2012).
- Investigation of two mammalian mRNAs revealed 3'-UTR G-quadruplexes that resulted in increased use of alternative polyadenylation sites. (See “Alternative cleavage, polyadenylation, and deadenylation” under “Creation of mRNA variants” above.) This has the effect of shortening the transcripts and therefore in turn regulating the regulation of the mRNA by microRNAs, which often target sites in the 3'-UTR. “Clearly, G-quadruplexes located in the 3'-UTRs of mRNAs are cis-regulatory elements that have a significant impact on gene expression” (Beaudoin and Perreault 2013).
- G-quadruplexes in 3'-UTRs of mRNA transcripts are only beginning to be explored. A recent study “revealed that a G4 [G-quadruplex] structure located in the 3'-UTR of two dendritic mRNAs can dictate their localization in neurites. Another one reported a G4 structure found in the 3'-UTR of the PIM1 mRNA acting as translational repressor. Moreover, RNA G4 structures have been reported to modulate the alternative splicing of the TP53 gene (encoding the p53 protein) and the hTERT gene (encoding the telomerase reverse transcriptase). In the case of the TP53 gene, an RNA G4 structure present downstream of the gene was reported to be crucial in maintaining an accurate 3'-end processing and function under conditions of stressing DNA damage” (Beaudoin and Perreault 2013).
- “RNA secondary structure is emerging as an important layer in splicing regulation. Here we demonstrate that RNA elements with G-quadruplex-forming capacity promote exon inclusion. Destroying G-quadruplex-forming capacity while keeping G tracts intact abrogates exon inclusion. Analysis of RNA-binding protein footprints revealed that G quadruplexes are enriched in heterogeneous nuclear ribonucleoprotein F (hnRNPF)-binding sites and near hnRNPF-regulated alternatively spliced exons in the human transcriptome. Moreover, hnRNPF regulates an epithelial–mesenchymal transition (EMT)-associated CD44 isoform switch in a G-quadruplex-dependent manner, which results in inhibition of EMT ... These data suggest a critical role for RNA G quadruplexes in regulating alternative splicing. Modulation of G-quadruplex structural integrity may control cellular processes important for tumor progression” (Huang, Zhang, Harvey et al. 2017a, doi:10.1101/gad.305862.117).
- “Polycomb repressive complex 2 (PRC2) interacts with chromatin to trimethylate histone H3 at Lys27 (H3K27me3) and repress gene expression, a process that is often dysregulated in cancer. Beltran et al. now reveal that PRC2–chromatin binding is regulated by chromatin-associated G-quadruplex (G4)-containing RNA, which binds PRC2, removes it from chromatin and reactivates gene expression” (Wrighton 2019, doi:10.1038/s41580-019-0184-5).
- See also “RNA structure” under “DECISION-MAKING DURING TRANSLATION” above, and “DNA G-quadruplexes” above.
-
Single-nucleotide “bulges”
Many mRNAs targeted by miRNAs for down-regulation do not quite match
the miRNA “seed” sequence that has been assumed to select the mRNA.
Instead, a single-nucleotide “bulge” in the mRNA plays an important
role during a two-stage match-up process between the mRNA and the
miRNA. The final result of the process is a match between six or more
nucleotides of the two RNAs, with the “bulge” nucleotide of the mRNA
sticking out from the duplex and not included in the match-up (Stefani
and Slack 2012, reporting on work by Chi, Hannon and Darnell).
- See one reported effect of such a bulge here.
-
Transient, dynamic conformation changes
RNAs shift between different conformations, including “excited states”
that “exist in too little abundance (2–13%) and for too short a
duration (45–250 ms) to allow structural characterization by
conventional techniques. Transitions towards ESs [excited states]
result in localized rearrangements in base-pairing that alter building
block elements of RNA architecture...The ES can inhibit function by
sequestering residues involved in recognition and signalling or promote
ATP-independent strand exchange. Thus, RNAs do not adopt a single
conformation, but rather exist in rapid equilibrium with alternative
ESs, which can be stabilized by cellular cues to affect functional
outcomes”. “We...predict that RNA ESs exist in great abundance
throughout the transcriptome” (Dethoff, Petzold, Chugh et al. 2012).
“RNAs fold into 3D structures that range from simple helical elements to
complex tertiary structures and quaternary ribonucleoprotein assemblies.
The functions of many regulatory RNAs depend on how their 3D structure
changes in response to a diverse array of cellular conditions. In this
Review, we examine how the structural characterization of RNA as dynamic
ensembles of conformations, which form with different probabilities and
at different timescales, is improving our understanding of RNA function
in cells. We discuss the mechanisms of gene regulation by microRNAs,
riboswitches, ribozymes, post-transcriptional RNA modifications and
RNA-binding proteins, and how the cellular environment and processes such
as liquid–liquid phase separation may affect RNA folding and activity.
The emerging RNA-ensemble–function paradigm is changing our perspective
and understanding of RNA regulation, from in vitro to in vivo and from
descriptive to predictive”
(Ganser, Kelly, Herschlag and Al-Hashimi 2019,
doi:10.1038/s41580-019-0136-0).
- “Compared to secondary structural transitions observed in many regulatory RNA switches, transitions between the ground and excited states uncovered here involve much more localized changes in RNA structure, occur at rates that are two-to-four orders of magnitude faster, and do not require assistance from external factors. Thus, they can meet unique demands in biological circuits and macro-molecular machines” (Dethoff, Petzold, Chugh et al. 2012). [The reader may or may not wish to excuse the gratuitous machine imagery.]
- “RNA dynamics play a fundamental role in many cellular functions. [There are] many structural maneuvers that occur over timescales ranging from picoseconds to seconds ... These transitions include large-scale secondary-structural transitions at >0.1-s timescales, basepair/tertiary dynamics at microsecond-to-millisecond timescales, stacking dynamics at timescales ranging from nanoseconds to microseconds, and other ‘jittering’ motions at timescales ranging from picoseconds to nanoseconds”. “RNAs often harness multiple modes to achieve complex functionality”. (Mustoe, Brooks and Al-Hashimi 2014, doi:10.1146/annurev-biochem-060713-035524)
- Human retinoblastoma is a cancer of the eye associated with the Retinoblastoma 1 (RB1) gene. The RB1 mRNA produced from this gene normally shifts dynamically between three distinct conformations. Two single-nucleotide mutations found in the RB1 5'-UTR of two retinoblastoma patients were shown to “collapse the structural ensemble to a single but distinct well-defined conformation”. The researchers conclude that “for the subset of patients with heritable retinoblastoma-associated single-nucleotide variations in the RB1 5′ UTR, the absence of multiple structures is likely causative of the cancer” (Kutchko, Sanders, Ziehr et al. 2015, doi:10.1261/rna.049221.114).
-
RNA 3' UTRs.
“3′ untranslated regions (3′ UTRs) of messenger RNAs (mRNAs) are best
known to regulate mRNA-based processes, such as mRNA localization, mRNA
stability, and translation. In addition, 3′ UTRs can establish 3′
UTR-mediated protein–protein interactions (PPIs), and thus can transmit
genetic information encoded in 3′ UTRs to proteins. This function has
been shown to regulate diverse protein features, including protein
complex formation or posttranslational modifications, but is also
expected to alter protein conformations. Therefore, 3′ UTR-mediated
information transfer can regulate protein features that are not encoded
in the amino acid sequence” (Mayr 2019, doi:11/10/a034728)
- Summary (mRNA only). “Increasing experimental and computational evidence points to the existence of extensive RNA structures in the coding regions of mRNA molecules. RNA secondary structures have been implicated in regulation of translation initiation, elongation and termination in both prokaryotes and eukaryotes. In particular, the anti-correlation between translation efficiency and the thermodynamic stability of local secondary structure in the vicinity of the translation initiation site has been thoroughly documented. RNA hairpins are thought to be involved in controlling mRNA decay, localization and interaction with other molecules. Overall, the mRNA coding regions appear to be more structured than the untranslated regions” (Chursov, Frishman and Shneider 2013, in a study of bacteria).
- See also “DNA-RNA triplexes” above.
-
Bioelectric effects
- In a dramatic video, Tufts University researchers show how the craniofacial (CF) structure of a developing frog tadpole is prefigured by the shifting biolectrical pattern in the region where the face will develop. They write that “ion flux directs cell behavior, it is not just a byproduct of cell physiology,” and “the locations of alkalinized/hyperpolarized cell domains overlap with the locations of CF developmental gene expression." In sum, "the pH and Vmem [membrane voltage] of these cells regulate expression of genes involved in CF [craniofacial] development” (Vandenberg, Morrie and Adams (2011).
-
Mechanical effects
Some key points on this topic:
- “Cellular mechanical states modulate cytoskeleton–nucleus links and trigger the translocation of regulatory molecules to the nucleus.
- “The remodelling of cytoskeleton–nucleus links results in distinct morphological as well as mechanical and dynamic properties of the cell nucleus.
- “The cell type-specific organization of chromosomes and their intermingling is modulated by the mechanical state of a cell.
- “The recruitment of transcription factors to their target genes is facilitated by the nuclear mechanical state through the establishment of particular chromosome neighbourhoods and functional gene clusters.
- “Such cell type-specific chromosome neighbourhoods and gene clusters are established during cell differentiation.
- “The spatial organization of chromosomes and their intermingling are crucial for mechanoregulation of gene expression, and alterations thereof can result in the onset of various diseases.”
“Cells generate and sense mechanical forces that trigger biochemical signals
to elicit cellular responses that control cell fate changes. Mechanical
forces also physically distort neighboring cells and the surrounding
connective tissue, which propagate mechanochemical signals over long
distances to guide tissue patterning, organogenesis, and adult tissue
homeostasis. As the largest and stiffest organelle, the nucleus is
particularly sensitive to mechanical force and deformation. Nuclear
responses to mechanical force include adaptations in chromatin architecture
and transcriptional activity that trigger changes in cell state. These
force-driven changes also influence the mechanical properties of chromatin
and nuclei themselves to prevent aberrant alterations in nuclear shape and
help maintain genome integrity”
(Miroshnikova and Wickström 2022, doi:10.1101/cshperspect.a039685).
- “Changes in the cytoskeleton originating from distant places like the cell membrane or even beyond generate forces that are transmitted to the nucleus resulting in changes in nuclear morphology and ultimately in changes in gene expression” (Castanon and González-Gaitán 2011). A key factor in this process is known as LINC (Linker of Nucleoplasm and Cytoskeleton), a protein complex that connects the lamina of the nuclear envelope with the cytoskeleton of the cytoplasm.
- “Mechanotransduction is a process whereby mechanical stimuli outside the cell are sensed by components of the plasma membrane and transmitted as signals through the cytoplasm that terminate in the nucleus. The nucleus responds to these signals by altering gene expression. During mechanotransduction, complex networks of proteins are responsible for cross talk between the cytoplasm and the nucleus. These proteins include cell membrane receptors, cytoplasmic filaments, LINC complex members that bridge the nucleus and cytoplasm, and nuclear envelope proteins that connect to the chromatin. Mechanotransduction also plays a critical role in development. Furthermore, it is possible that disrupted mechanotransduction leads to changes in gene expression that underlie the pathogenic mechanisms of disease” (Wallrath, Bohnekamp and Magin 2016, doi:10.1016/j.gde.2016.03.007).
- “Mechanical coupling of the nucleus with the cytoskeleton plays a critical role in physiological processes, such as nuclear positioning during cell migration and provides a protein network capable of transferring environmental signals to the nucleus to activate new transcriptional programs” (Berger and Geyer 2016, doi:10.1016/j.gde.2016.03.007).
- “We show that a mechanosensory complex of emerin (Emd), non-muscle myosin IIA (NMIIA) and actin controls gene silencing and chromatin compaction, thereby regulating lineage commitment. Force-driven enrichment of Emd at the outer nuclear membrane of epidermal stem cells leads to defective heterochromatin anchoring to the nuclear lamina and a switch from H3K9me2,3 to H3K27me3 occupancy at constitutive heterochromatin. Emd enrichment is accompanied by the recruitment of NMIIA to promote local actin polymerization that reduces nuclear actin levels, resulting in attenuation of transcription and subsequent accumulation of H3K27me3 at facultative heterochromatin. Perturbing this mechanosensory pathway by deleting NMIIA in mouse epidermis leads to attenuated H3K27me3-mediated silencing and precocious lineage commitment, abrogating morphogenesis” (Le, Ghatak, Yeung et al. 2016, doi:10.1038/ncb3387).
- “Mechanical strain, transmitted by the remodelling of the actomyosin cytoskeleton and concomitant depletion of the nuclear actin pool, is shown to induce silencing of differentiation genes in epidermal stem cells, linking mechanical cues to the genetic regulation of cell fate” (table of contents summary for Strzyz 2016, doi:10.1038/nrm.2016.105).
- “Cells are subject to many mechanical stresses. These forces can profoundly alter cellular biochemistry. Tajik et al. used a magnetic bead on the surface of a cell to apply a shear stress to the cell's nucleus. A linear array of fluorophoremarked genes moved apart, indicating that their chromatin was being stretched. Stretching activates transcription of the marker genes within seconds, suggesting that the stretching effect, propagated through cytoskeletal tension, is direct, with the degree of stretching correlating with the degree of gene activation” (Riddihough 2016 [Science, Sep. 23, p. 1378], reporting on doi:10.1038/NMAT4729).
-
Regarding “mechanotransduction” of external forces to the nucleus:
“Chromosome intermingling regions are mechanical hotspots for genome
regulation. Maintenance of such mechanical hotspots is crucial for
cellular homeostasis, and alterations in them could be precursors for
various cellular reprogramming events, including diseases”. In
particular:
- “Microenvironment signals are transmitted to the cell nucleus via both physical and biochemical intermediates.
- “The spatial organization of chromosomes is critical to regulating microenvironmental control of gene expression.
- “Intermingling regions between chromosomes are enriched with transcription factors and RNA Pol II.
- “The functional clustering of genes is modulated by microenvironmental signals to exhibit differential gene expression programs”
- “Mechanotransduction — the process of converting mechanical forces into biochemical signals — often terminates in the nucleus, leading to changes in gene expression. These changes in gene expression can be achieved by mechanosensitive transcription factors, such as the effector of Hippo signalling pathway Yes-associated protein (YAP), which translocates to the nucleus in a manner that is regulated by extracellular mechanical signals. The exact mechanisms that govern the import of proteins into the nucleus in response to mechanical stimuli remain poorly understood. Roca-Cusachs and colleagues now show that the key event regulating this shuttling is the application of force to the nucleus, which promotes nuclear import by reducing the permeability barrier of nuclear pores” (Strzyz 2017, doi:10.1038/nrm.2017.114).
-
Phase transitions and membraneless organelles
“A number of studies have shown that membrane-less assemblies exhibit
remarkable liquid-like features. As with conventional liquids, they
typically adopt round morphologies and coalesce into a single droplet upon
contact with one another and also wet intracellular surfaces such as the
nuclear envelope. Moreover, component molecules exhibit dynamic exchange
with the surrounding nucleoplasm and cytoplasm. These findings together
suggest that these structures represent liquid-phase condensates, which form
via a biologically regulated (liquid-liquid) phase separation process.
Liquid phase condensation increasingly appears to be a fundamental mechanism
for organizing intracellular space. Consistent with this concept, several
membrane-less organelles have been shown to exhibit a concentration
threshold for assembly, a hallmark of phase separation. At the molecular
level, weak, transient interactions between molecules with multivalent
domains or intrinsically disordered regions (IDRs) are a driving force for
phase separation. In cells, condensation of liquid-phase assemblies can be
regulated by active processes, including transcription and various
posttranslational modifications. The simplest physical picture of a
homogeneous liquid phase is often not enough to capture the full complexity
of intracellular condensates, which frequently exhibit heterogeneous
multilayered structures with partially solid-like characters. However,
recent studies have shown that multiple distinct liquid phases can coexist
and give rise to richly structured droplet architectures determined by the
relative liquid surface tensions. Moreover, solid-like phases can emerge
from metastable liquid condensates via multiple routes of potentially both
kinetic and thermodynamic origins, which has important implications for the
role of intracellular liquids in protein aggregation pathologies”
(Shin and Brangwynne 2017, doi:10.1126/science.aaf4382).
“Formation of most BioMCs [biomolecular concentrates (=membraneless
organelles)] is governed by phase separation of high local concentrations of
multivalent molecular assemblies including multi‐domain proteins, proteins
harboring intrinsically disordered domains or prion‐like domains, RNA
recognition motifs, and RNA molecules. Various interactions between
proteins and RNAs contribute to the material state and behavior of BioMCs
including the constant exchange of molecules with the surrounding liquid
phase. Disruption of these interactions and dissolution of BioMCs can be
brought about through activities that target protein content including
post‐translational modifications (PTMs), chaperones or specific protein
degradation, or that modulate RNA content including competition by
additional RNAs, helicases, nucleases, or post‐transcriptional
modifications.
“RNA can serve as ‘trigger’, aggregating BioMCs, as ‘glue’, scaffolding BioMCs, as ‘exchange material’ that associates with established BioMCs, and potentially as ‘access point’ for activities changing BioMC architecture and possibly dissolving BioMCs ... Direct molecular ‘manipulation’ of expressed RNA molecules and proteins is likely the means for achieving the dynamic regulation of the multitude of non‐membranous organelles” (Drino and Schaefer 2018, doi:10.1002/bies.201800085).
“The recent report of molecular condensation properties of AGO and TNRC6
connects miRNA regulation to the growing field of biological phase
separation. The data demonstrate that miRISCs can form large molecular
condensates in vitro and in living cells, and it was hypothesized that the
ability to form higher order complexes through molecular condensation may
allow miRISCs to organize miRNA–target interactions within the cytoplasm and
thereby modulate rates of mRNA translation and decay. This hypothesis raises
the possibility that miRNA activity is regulated through the assembly of the
miRISC itself by modulation of the biophysical properties of miRISC
components”
(Gebert and MacRae 2018, doi:10.1038/s41580-018-0045-7).
“Cells under stress must adjust their physiology, metabolism, and
architecture to adapt to the new conditions. Most importantly, they must
down-regulate general gene expression, but at the same time induce synthesis
of stress-protective factors, such as molecular chaperones ... [We] propose
that the solubility of important translation factors is specifically
affected by changes in physical–chemical parameters such temperature or pH
and modulated by intrinsically disordered prion-like domains. These
stress-triggered changes in protein solubility induce phase separation into
condensates that regulate the activity of the translation factors and
promote cellular fitness. Prion-like domains play important roles in this
process as environmentally regulated stress sensors and modifier sequences
that determine protein solubility and phase behavior”
(Franzmann and Alberti 2019, doi:10.1101/cshperspect.a034058).
“Recent structural studies have elucidated mechanisms that govern the
regulation of transcription by RNA polymerases during the initiation and
elongation phases. Microscopy studies have revealed that transcription
involves the condensation of factors in the cell nucleus. A model is
emerging for the transcription of protein-coding genes in which distinct
transient condensates form at gene promoters and in gene bodies to
concentrate the factors required for transcription initiation and
elongation, respectively. The transcribing enzyme RNA polymerase II may
shuttle between these condensates in a phosphorylation-dependent manner.
Molecular principles are being defined that rationalize transcriptional
organization and regulation, and that will guide future investigations”
(Cramer 2019, doi:10.1038/s41586-019-1517-4).
“Eukaryotic chromatin is highly condensed but dynamically accessible to
regulation and organized into subdomains. We demonstrate that reconstituted
chromatin undergoes histone tail-driven liquid-liquid phase separation
(LLPS) in physiologic salt and when microinjected into cell nuclei,
producing dense and dynamic droplets. Linker histone H1 and internucleosome
linker lengths shared across eukaryotes promote phase separation of
chromatin, tune droplet properties, and coordinate to form condensates of
consistent density in manners that parallel chromatin behavior in cells.
Histone acetylation [which is generally supportive of gene expression] by
p300 antagonizes chromatin phase separation, dissolving droplets in vitro
and decreasing droplet formation in nuclei. In the presence of
multi-bromodomain proteins, such as BRD4, highly acetylated chromatin forms
a new phase-separated state with droplets of distinct physical properties,
which can be immiscible with unmodified chromatin droplets, mimicking
nuclear chromatin subdomains. Our data suggest a framework, based on
intrinsic phase separation of the chromatin polymer, for understanding the
organization and regulation of eukaryotic genomes”
(Gibson, Doolittle, Schneider et al. 2019, doi:10.1016/j.cell.2019.08.037).
“Genome expression and stability are dependent on biological processes that
control repetitive DNA sequences and nuclear compartmentalization. The phase
separation of macromolecules has recently emerged as a major player in the
control of biological pathways. Here, we summarize recent studies that
collectively reveal intersections between phase separation, repetitive DNA
elements, and nuclear compartments. These intersections modulate fundamental
processes, including gene expression, DNA repair, and cellular lifespan, in
the context of health and diseases such as cancer and neurodegeneration”
(abstract of article, “Phase Separation as a Melting Pot for DNA Repeats” by
Hall, Ostrowski and Mekhail 2019, doi:10.1016/j.tig.2019.05.001).
“Chromatin readers are important intermediaries linking epigenetic
information and biological phenotypes. Many diseases are caused by mutations
in epigenetic readers. Recently, a study by Wan et al. uncovered that
cancer-associated mutations promote self-association of
eleven-nineteen-leukemia protein (ENL), leading to abnormal condensates,
elevated gene expression, and impaired cell fate determination”. “It is
tempting to hypothesize that the pathogenic effect of ENL mutants is likely
due to gain-of-LLPS [liquid-liquid phase separation] properties, which
elevates transcription of target genes”
(Gao and Li 2020, doi:10.1016/j.tibs.2020.02.007).
“Expansions of amino acid repeats occur in >20 inherited human disorders,
and many occur in intrinsically disordered regions (IDRs) of transcription
factors (TFs). Such diseases are associated with protein aggregation, but
the contribution of aggregates to pathology has been controversial. Here, we
report that alanine repeat expansions in the HOXD13 TF, which cause
hereditary synpolydactyly in humans, alter its phase separation capacity and
its capacity to co-condense with transcriptional co-activators. HOXD13
repeat expansions perturb the composition of HOXD13-containing condensates
in vitro and in vivo and alter the transcriptional program in a
cell-specific manner in a mouse model of synpolydactyly. Disease-associated
repeat expansions in other TFs (HOXA13, RUNX2, and TBP) were similarly found
to alter their phase separation. These results suggest that unblending of
transcriptional condensates may underlie human pathologies”
(Basu, Mackowiak, Niskanen, et al. 2020, doi:10.1016/j.cell.2020.04.018).
“Cellular functioning requires the orchestration of thousands of molecular
interactions in time and space. Yet most molecules in a cell move by
diffusion, which is sensitive to external factors like temperature. How
cells sustain complex, diffusion-based systems across wide temperature
ranges is unknown. Here, we uncover a mechanism by which budding yeast
modulate viscosity in response to temperature and energy availability. This
‘viscoadaptation’ uses regulated synthesis of glycogen and trehalose to
vary the viscosity of the cytosol. Viscoadaptation functions as a stress
response and a homeostatic mechanism, allowing cells to maintain invariant
diffusion across a 20C temperature range. Perturbations to viscoadaptation
affect solubility and phase separation, suggesting that viscoadaptation
may have implications for multiple biophysical processes in the cell.
Conditions that lower ATP trigger viscoadaptation, linking energy
availability to rate regulation of diffusion-controlled processes.
Viscoadaptation reveals viscosity to be a tunable property for regulating
diffusion-controlled processes in a changing environment”
(Persson, Ambati and Brandman 2020, doi:10.1016/j.cell.2020.10.017).
“Regulation of transcription is a fundamental cellular process where the
mechanisms involved in initiation have been studied extensively, but those
involved in arresting the process are poorly understood. Modeling of the
potential roles of RNA in transcriptional control suggested a
non-equilibrium feedback control mechanism where low levels of RNA promote
condensates formed by electrostatic interactions whereas relatively high
levels promote dissolution of these condensates. Evidence from in
vitro and in vivo experiments support a model where RNAs produced
during early steps in transcription initiation stimulate condensate
formation, whereas the burst of RNAs produced during elongation stimulate
condensate dissolution. We propose that transcriptional regulation
incorporates a feedback mechanism whereby transcribed RNAs initially
stimulate but then ultimately arrest the process.” “Charge balance of
electrostatic interactions can account for RNA feedback regulation”
(Henninger, Oksuz, Shrinivas et al. 2021, doi:10.1016/j.cell.2020.11.030).
“Dynamic morphological changes of intracellular organelles are often
regulated by protein phosphorylation or dephosphorylation. Phosphorylation
modulates stereospecific interactions among structured proteins, but how it
controls molecular interactions among unstructured proteins and regulates
their macroscopic behaviours remains unknown. Here we determined the cell
cycle-specific behaviour of Ki-67, which localizes to the nucleoli during
interphase and relocates to the chromosome periphery during mitosis. Mitotic
hyperphosphorylation of disordered repeat domains of Ki-67 generates
alternating charge blocks in these domains and increases their propensity
for liquid–liquid phase separation (LLPS). A phosphomimetic sequence and the
sequences with enhanced charge blockiness underwent strong LLPS in vitro and
induced chromosome periphery formation in vivo. Conversely, mitotic
hyperphosphorylation of NPM1 diminished a charge block and suppressed LLPS,
resulting in nucleolar dissolution. Cell cycle-specific phase separation can
be modulated via phosphorylation by enhancing or reducing the charge
blockiness of disordered regions, rather than by attaching phosphate groups
to specific sites”
(Yamazaki, Takagi, Kosako et al. 2022, doi:10.1038/s41556-022-00903-1)
“Membraneless organelles (MLOs) are detected in cells as dots of mesoscopic
size. By undergoing phase separation into a liquid-like or gel-like phase,
MLOs contribute to intracellular compartmentalization of specific biological
functions. In eukaryotes, dozens of MLOs have been identified, including the
nucleolus, Cajal bodies, nuclear speckles, paraspeckles, promyelocytic
leukaemia protein (PML) nuclear bodies, nuclear stress bodies, processing
bodies (P bodies) and stress granules. MLOs contain specific proteins, of
which many possess intrinsically disordered regions (IDRs), and nucleic
acids, mainly RNA. Many MLOs contribute to gene regulation by different
mechanisms. Through sequestration of specific factors, MLOs promote
biochemical reactions by simultaneously concentrating substrates and
enzymes, and/or suppressing the activity of the sequestered factors
elsewhere in the cell. Other MLOs construct inter-chromosomal hubs by
associating with multiple loci, thereby contributing to the biogenesis of
macromolecular machineries essential for gene expression, such as ribosomes
and spliceosomes. The organization of many MLOs includes layers, which might
have different biophysical properties and functions. MLOs are functionally
interconnected and are involved in various diseases, prompting the emergence
of therapeutics targeting them. In this Review, we introduce MLOs that are
relevant to gene regulation and discuss their assembly, internal structure,
gene-regulatory roles in transcription, RNA processing and translation,
particularly in stress conditions, and their disease relevance”
(Hirose, Ninomiya, Nakagawa and Yamazaki 2023,
doi:10.1038/s41580-022-00558-8)
“Biomolecular condensates are recognized for their ability to
compartmentalize the cytoplasm without bounding membranes, but the degree to
which they organize the cytoplasm has not been clear. A new study reveals
that condensates at a scale of 100 nm are responsible for the organization
of at least 18% of the cytoplasmic proteome”
(Liyanage and Ditlev 2024, doi:10.1038/s41556-023-01331-5).
- “Remarkably simple proteins play outsize roles in the execution of developmental complexity within biological systems. Sequence information determines structure and hence function, so how do low complexity sequences fulfill their functions? Recent discoveries are raising the curtain on a new dimension of the sequence-structure paradigm. In it, function derives not from the structures of individual proteins, but instead, from dynamic material properties of entire ensembles of the proteins acting in unison through phase changes. These phases include liquids, one-dimensional crystals, and — as elaborated herein — even glasses. The peculiar thermodynamics of glass-like protein assemblies, in particular, illuminate new principles of information flow through and, at times, orthogonal to the central dogma of molecular biology” (Halfmann 2016, doi:10.1016/j.sbi.2016.05.002).
- “How, with an aversion to structure, do IDRs [intrinsically disordered regions] assemble some of the largest structures in the cell? The answer is deceptively simple. As if for water vapor condensing into dew droplets, the proteins coalesce out of the bulk cellular milieu into their own liquid phases. Unlike structured macromolecular complexes, individual polypeptides remain disordered within the liquid protein droplets, fleeting between self-solvated, energetically comparable intermolecular conformations. Attached globular domains and interacting macromolecules are pulled in along with their disordered partners. The compartmentalization of proteins within the liquid phase facilitates regulatory processes, including signaling, transcription, mRNA processing, and nucleation of cytoskeletal polymers. The droplet-dependent localization of these activities enforces cell polarity, symmetry breaking, and cell differentiation. Intriguingly, droplets have been observed to solidify, or ‘mature’, over time, and this may be a basis for both functional and pathological differentiation of droplet activities” (Halfmann 2016, doi:10.1016/j.sbi.2016.05.002).
- New research shows that “low complexity sequences and intrinsically disordered regions in splicing factors induce the formation of higher-order protein assemblies” by means of liquid-liquid phase transitions”. This process is thought to “concentrate and bring together different splicing components” and therefore to help regulate alternative splicing (Zlotorynski 2017, doi:10.1038/nrm.2017.78).
- “Cells compartmentalize biochemical reactions using organelles. Organelles can be either membrane-bound compartments or supramolecular assemblies of protein and ribonucleic acid known as ‘biomolecular condensates’. Biomolecular condensates, such as nucleoli and germ granules, have been described as liquid like, as they have the ability to fuse, flow, and undergo fission. Recent experiments have revealed that some liquid-like condensates can mature over time to form stable [bioactive] gels. In other cases, biomolecular condensates solidify into amyloid-like fibers ... the material properties of these condensates can be explained by the principles of liquid–liquid phase separation and maturation” (Woodruff, Hyman and Boke 2018, doi:10.1016/j.tibs.2017.11.005).
- “Transcription factors, including ERα [estrogen receptor alpha], downstream co-activators, and RNA polymerase II, form biomolecular condensates in vivo at their respective super-enhancer target sites by a mechanism known as ‘phase separation’. Importantly, condensate formation was shown to be required for super-enhancer function and gene activation” (Wittmann and Alberti 2019, doi:10.1038/s41594-019-0198-x).
- “The long non-coding RNA Xist induces heterochromatinization of the X chromosome by recruiting repressive protein complexes to chromatin. Here we gather evidence, from the literature and from computational analyses, showing that Xist assemblies are similar in size, shape and composition to phase-separated condensates, such as paraspeckles and stress granules. Given the progressive sequestration of Xist’s binding partners during X-chromosome inactivation, we formulate the hypothesis that Xist uses phase separation to perform its function” (Cerase, Armaos, Neumayer et al. 2019, doi:10.1038/s41594-019-0223-0).
-
“Parentally deposited small non-coding RNAs direct heritable gene
regulation in the C. elegans germline. Dodson and Kennedy provide
evidence that biomolecular condensates known as germ granules spatially
organize these small RNA-based epigenetic inheritance pathways.
Disrupting germ granules triggers changes in small-RNA-based gene
regulation that can be inherited across generations”
(table of contents blurb for Dodson and Kennedy 2019,
doi:10.1016/j.devcel.2019.07.025).
And another toc blurb: “Germ granules are perinuclear condensates in germ cells. Ouyang et al. report that germ (P) granules in C. elegans harbor transcripts required for RNA-mediated interference. Localization to P granules protects these transcripts from piRNA-initiated silencing, identifying a mechanism for regulating RNAi responses in animals” (Ouyang, Folkmann, Bernard et al. 2019, doi:10.1016/j.devcel.2019.07.026).
- “Changes in RNA abundance in transcriptional condensates provide dynamic feedback on transcription.” “RNA may provide first positive and then negative feedback on transcription, by regulating electrostatic interactions in transcriptional condensates” (Zlotorynski 2021, doi:10.1038/s41580-021-00340-2).
- “Cellular condensates such as granules are generally in a liquid state, but can solidify as they ‘age’; the significance of this solid state is largely unknown. Bose at al. now report that granules in fruit flies containing the developmental mRNA oskar transition to a solid state, which is required for localized translation and embryogenesis” (Zlotorynski 2022, doi:10.1038/s41580-022-00477-8).
-
Ribonucleoprotein phase transitions
mRNAs typically associate with various proteins, forming
ribonucleoprotein (RNP) complexes. As some sections of these notes
testify, RNA-protein interactions are crucial to the regulation of gene
expression. But there is now emerging a more fundamental truth not
otherwise touched on here: large-scale, macromolecular assemblies of
RNPs can form liquid, semi-liquid, or solid aggregates within the cell,
and regulated phase transitions among these states are important for
life processes (Hubstenberger, Noble, Cameron and Evans 2013; Hyman and
Brangwynne 2011).
“The cell nucleus contains a large number of membrane-less bodies that
play important roles in the spatiotemporal regulation of gene expression.
Recent work suggests that low complexity/disordered protein motifs and
repetitive binding domains drive assembly of droplets of nuclear
RNA/protein by promoting nucleoplasmic phase separation. Nucleation and
maturation of these structures is regulated by, and may in turn affect,
factors including post-translational modifications, protein
concentration, transcriptional activity, and chromatin state” (Zhu and
Brangwynne 2015, doi:10.1016/j.ceb.2015.04.003).
“Living cells organize functions not only by membrane
compartmentalization, but also by assembling supramolecular structures
within aqueous environments. ... Supramolecular assemblies are emerging
as a prominent feature of gene expression pathways. ... Specific RNPs
often coassemble into a remarkable diversity of large RNP granules or
domains. In the nucleus, these structures include the nucleolus, Cajal
bodies, and a variety of other nuclear RNP particles. Diverse RNP
assemblies are also common in the cytoplasm and include P-bodies,
stress granules, neuronal granules, U-bodies, germ granules, and a
variety of PB/SG-related granule types that form in early development.
Like chromatin, these RNP assemblies are regulated by developmental
programs and cell state changes, suggesting important roles in
controlling cell fates” (Hubstenberger, Noble, Cameron and Evans 2013).
“Regarding the phase separation of proteins as liquid droplets in the
cytoplasm: “It turned out that especially low-complexity
RBPs [RNA-binding proteins] such as FUS (Fused in Sarcoma), hnRNPA1 and
hnRNPA2B1 were abundant in the droplets/hydrogels. Intriguingly, mRNAs
in the hydrogels exhibited a preponderance of long 3'UTRs, which provide
a platform for RBP interplay, and could mean that mRNAs in liquid
droplets are likely to be regulated mRNA species. An emerging theme in
mRNA studies is that extensive 3'UTRs act as scaffolds for
post-transcriptional regulatory purposes, and that physiologic responses
depend on competing and/or cooperating trans-acting factors”. There is
complex interaction “between multiple RNA-binding modules in RBPs,
multiple RBP target sites on RNA, and multiple low-complexity sequences
in RBPs”. “Because a single RBP may literally have thousands of
different interactions with the transcriptome, and hundreds of RBPs may
behave in this manner, the complexity is daunting. Nevertheless, the
combinatorial binding or competition among RBPs determines the fate of
the individual transcript”
(Nielsen, Hansen and Christiansen 2016, 10.1002/bies.201500175).
“Phase separation is emerging as a paradigm to explain the self-assembly
and organization of membraneless bodies in the cell. Recent advances show
that this principle also extends to nucleoprotein complexes, including
DNA-based structures. We discuss here recent observations on the role of
phase separation in genome organization across the evolutionary spectrum
from bacteria to mammals. These findings suggest that molecular
interactions amongst DNA-binding proteins evolved to form a variety of
biomolecular condensates with distinct material properties that affect
genome organization and function. We suggest that phase separation
contributes to genome organization across evolution and that the
resulting phase behavior of genomes may underlie regulatory mechanisms
and disease”
(Feric and Misteli 2021, doi:10.1016/j.tcb.2021.03.001)
“DEAD-box ATPases constitute a very large protein family present in all
cells, often in great abundance. From bacteria to humans, they play
critical roles in many aspects of RNA metabolism, and due to their
widespread importance in RNA biology, they have been characterized in
great detail at both the structural and biochemical levels. DEAD-box
proteins function as RNA-dependent ATPases that can unwind short duplexes
of RNA, remodel ribonucleoprotein (RNP) complexes, or act as clamps to
promote RNP assembly. Yet, it often remains enigmatic how individual
DEAD-box proteins mechanistically contribute to specific RNA-processing
steps. Here, we review the role of DEAD-box ATPases in the regulation of
gene expression and propose that one common function of these enzymes is
in the regulation of liquid–liquid phase separation of RNP condensates”
(Weis and Hondele 2022, doi:10.1146/annurev-biochem-032620-105429).
- Hubstenberger et al. (2013) looked at early developmental processes in the nematode, Caenorhabditis elegans. “Here we show that regulated RNP factor interactions drive transitions among diffuse, semiliquid, or solid states to modulate RNP sorting and exchange in the Caenorhabditis elegans oocyte cytoplasm”. Further, “RNP phase transitions are controlled with surprising precision in early development, leading to starkly different supramolecular states that alter RNP organization and dynamics. ... Pathways of mRNA regulation control these transitions. ... Reversible interactions among thousands of RNP complexes impart regulated patterns of RNP dynamics, and large-scale organization of gene expression pathways in the cytoplasm”.
- More particularly: during early C. elegans development, “translation repressors induce an intrinsic capacity of RNP components to coassemble into either large semiliquids or solid lattices, whereas a conserved RNA helicase prevents polymerization into nondynamic solids. Developmental cues dramatically alter both fluidity and sorting within large RNP assemblies, inducing a transition from RNP segregation in quiescent oocytes to dynamic exchange in the early embryo”. (Hubstenberger, Noble, Cameron and Evans 2013).
- “In an exciting recent advance, it has been demonstrated that low-affinity, multivalent protein–protein interactions, often mediated by low-complexity and prion-like intrinsically disordered sequences, can promote liquid–liquid demixing to form membrane-less cytoplasmic and nuclear granules. These granules behave like dynamic liquid droplets, rapidly exchanging component proteins and RNA with the cytoplasm or nucleoplasm. By sequestering regulatory proteins under conditions of high macromolecular concentration, intrinsically disordered protein-mediated phase separation can have a profound influence on cellular signalling” (Wright and Dyson 2015, doi:10.1038/nrm3920). Through their roles both in signaling and in the phase separation that sustains membrane-less nuclear “bodies”, these disordered proteins have profound (and certainly non-codelike!) influences on gene expression.
- “Phase changes can be a strong function of salt, protein concentration, and temperature” (Zhu and Brangwynne 2015, doi:10.1016/j.ceb.2015.04.003).
- “A key question is how dynamic low-affinity interactions can give rise to compositionally specified bodies. Why do some proteins localize to only the nucleolus, while others can be found in both the nucleolus and Cajal bodies? Can we begin to construct multi-dimensional phase diagrams that specify the molecular concentrations and degree of post-translational modification that promote assembly of various types of nuclear bodies ...? How is the logic of molecular specificity encoded in the promiscuous interactions of intrinsically disordered proteins? ... A central challenge is to elucidate what increasingly appears to be an intimate, but still poorly understood, feedback between nuclear body assembly/disassembly, chromatin compaction state, transcriptional activity, and RNA processing” (Zhu and Brangwynne 2015, doi:10.1016/j.ceb.2015.04.003).
- “Currently increasing evidence suggests that chromatin has a dynamic liquid-like structure based on the 10-nm fiber but not the 30-nm fiber. This liquid-like property can drive the process of ‘scanning and targeting genomic DNA,’ which contributes to various genome functions including gene expression and DNA replication, repair, and recombination” (Maeshima, Ide, Hibino and Sasai 2016, doi:10.1016/j.gde.2015.11.006).
- “In eukaryotic cells, diverse stresses trigger coalescence of RNA-binding proteins into stress granules. In vitro, stress-granule-associated proteins can demix to form liquids, hydrogels, and other assemblies lacking fixed stoichiometry. Observing these phenomena has generally required conditions far removed from physiological stresses. We show that poly(A)-binding protein (Pab1 in yeast), a defining marker of stress granules, phase separates and forms hydrogels in vitro upon exposure to physiological stress conditions. Other RNA-binding proteins depend upon low-complexity regions (LCRs) or RNA for phase separation, whereas Pab1’s LCR is not required for demixing, and RNA inhibits it ... Mutations that impede phase separation reduce organism fitness during prolonged stress. Poly(A)-binding protein thus acts as a physiological stress sensor, exploiting phase separation to precisely mark stress onset, a broadly generalizable mechanism” (Riback, Katanski, Kear-Scott et al. 2017, doi:10.1016/j.cell.2017.02.027).
- “miRISC is a multi-protein assembly that uses microRNAs (miRNAs) to identify mRNAs targeted for repression. Dozens of miRISC-associated proteins have been identified, and interactions between many factors have been examined in detail ... Here, we show that two core protein components of human miRISC, Argonaute2 (Ago2) and TNRC6B, condense into phase-separated droplets in vitro and in live cells. Phase separation is promoted by multivalent interactions between the glycine/tryptophan (GW)-rich domain of TNRC6B and three evenly spaced tryptophan-binding pockets in the Ago2 PIWI domain. miRISC droplets formed in vitro recruit deadenylation factors and sequester target RNAs from the bulk solution. The condensation of miRISC is accompanied by accelerated deadenylation of target RNAs bound to Ago2. The combined results may explain how miRISC silences mRNAs of varying size and structure and provide experimental evidence that protein-mediated phase separation can facilitate an RNA processing reaction” (Sheu-Gruttadauria and MacRae 2018, doi:10.1016/j.cell.2018.02.051).
- “The interplay between LLPS [liquid-liquid phase separation] and chromatin is thus able to generate significant forces that can both push chromatin regions away from each other as well as bring them together. This phenomenon demonstrates a new highly versatile potential mechanism for controlling the 3D arrangement of regulatory elements, aspects which are likely to be key for chromosomal communication and thus cell differentiation and gene expression” (Welsh, Shen, Levin and Knowles 2018, doi:10.1016/j.cell.2018.11.020).
-
Structured water
Consider this item to be a placeholder for future content. I have long
thought that some day water will be seen as the most fundamentally
important, “information-rich” biomolecule of all and that revelations
in this regard will outweigh in significance even those concerning the
structure of the double helix. No biologist today would suggest such a
thing — and I am not defending the idea here, if only for lack of
ability. I am content to let time decide the matter. Meanwhile,
however, you might find
this
brief passage of interest.
-
Genome remodeling
As molecular biologists begin to sequence the genomes of multiple tissues
within a single individual, they are discovering the kind of variation that
led one researcher (Lupski 2013) to write an article in Science
entitled “Genome Mosaicism — One Human, Multiple Genomes”. That is, our
bodies are mosaics of cell populations, each with a distinct genome due to
many sorts of alterations that occur during development. “It is becoming
increasingly apparent that a human individual is made up of a population of
cells, each with its own ‘personal’ genome”. And so modifying its own
genome becomes one of the most fundamental ways that the organism can
manage its gene expression.
This topic is also dealt with in the sections,
“Extrachromosomal DNA” and
“Retrotransposons”.
The “canonical” source on this topic — which can embrace vastly more than is
discussed here — is University of Chicago microbiologist James Shapiro. See
his book, Evolution: A View from the 21st Century.
For example, Tables II.8, II.11, and III.2 at the website contain many
useful references. (Shapiro uses the phrase, “natural genetic
engineering”.)
From the “Highlights” section of Campbell, Shaw, Stankiewicz and Lupski
2015, doi:10.1016/j.tig.2015.03.013:
“Postzygotic mutation is a common occurrence. The developmental stage and timing of new mutations influence their phenotypic effects and likelihood of transmission. All major classes of mutations are observed in the mosaic state”.
And from their abstract: “Nearly all of the genetic material among cells within an organism is identical. However, single-nucleotide variants, small insertions/deletions, copy-number variants, and other structural variants continually accumulate as cells divide during development. This process results in an organism composed of countless cells, each with its own unique personal genome. Thus, every human is undoubtedly mosaic. Mosaic mutations can go unnoticed, underlie genetic disease or normal human variation, and may be transmitted to the next generation as constitutional variants”.
- It appears that 30% of human skin fibroblast cells “have somatic copy number variations [CNVs] in their genomes, suggesting widespread somatic mosaicism in the human body” (Abyzov, Mariani, Palejev et al. 2012).
- “Dramatic genome dynamics, such as chromosome instability, contribute to the remarkable genomic heterogeneity among the blastomeres comprising a single embryo during human preimplantation development. This heterogeneity, when compatible with life, manifests as constitutional mosaicism, chimerism, and mixoploidy in live-born individuals. Chimerism and mixoploidy are defined by the presence of cell lineages with different parental genomes or different ploidy states in a single individual, respectively ... We not only demonstrate that chromosome instability is conserved between bovine and human cleavage embryos, but we also discovered that zygotes can spontaneously segregate entire parental genomes into different cell lineages during the first post-zygotic cleavage division. Parental genome segregation was not exclusively triggered by abnormal fertilizations leading to triploid zygotes, but also normally fertilized zygotes can spontaneously segregate entire parental genomes into different cell lineages during cleavage of the zygote” (Destouni, Esteki, Catteeuw et al. 2016, doi:10.1101/gr.200527.115).
- Aneuploidy and CNVs in the brain: (an extra or missing chromosome), according to Bushman and Chun (2013,) occurs in 30–35% of cells in the developing human brain. And while the amount at maturity isn’t yet known, “a significant population of aneuploid cells is also present in the adult human brain” — about 10% of cells by some estimates. “Studies of aneuploidy in the non-diseased central nervous system question the assumption that aneuploidy is in fact ‘abnormal’ in the development of certain cell lineages, and that it is deleterious — views contradicted by the maintenance of aneuploid populations in the normal brain. ... The stable and seemingly permanent changes produced by genomic alterations in a single neuron could provide a mechanism for creating and stabilizing functional mosaic populations within the brain, such as those constituting a neural network”.
- Looking more generally at the many types of genomic variation in the brain (not only aneuploidy), one sees regional variations between the frontal cortex and cerebellum within a single individual, suggesting that there are “non-random mechanisms in the generation and/or maintenance of this variability” (Bushman and Chun 2013).
- Another study, unlike others, was designed to pick up large-scale (greater than 1 megabase) copy number variations (CNVs) in neuronal genomes. “Single-cell sequencing of endogenous human frontal cortex neurons revealed that 13 to 41% of neurons have at least one megabase-scale de novo CNV [copy number variation], that deletions are twice as common as duplications, that a subset of neurons have highly aberrant genomes, marked by multiple alterations”. “These results demonstrate that somatic copy number variations are a common feature of neuronal genomes and suggest that the relative abundance of different CNV classes may vary among individuals” (McConnell, Lindberg, Brennand et al. 2013).
- Embryonic development: It is now understood that the early cell divisions of the human embryo produce massive amounts of DNA variation, with one group reporting chromosomal changes in a “staggering 70%” of the embryos examined. They found “not only mosaicism for whole-chromosome aneuploidies” in most of the embryos, “but also frequent segmental deletions, duplications and amplifications”. They concluded that “chromosome instability is prevalent in human embryogenesis” (Vanneste, Voet and Le Caignec 2009). Based on the study of both in vitro and in vivo pregnancies, “it is now clear that structural aberrations are a common occurrence in preimplantation embryos” (Mertzanidou et al. 2013). However, these “aberrations”, as we heard from Bushman and Chun above, are looking more and more like part of the intentional activity of the organism.
-
Polyploidy and aneuploidy in general.
Although “substantial differences in DNA content have been observed in
many human cell types”, such observations “have done little to change the
traditional view that all healthy somatic cells in the human body hold
the same characteristic quantity of DNA”. Nevertheless, New “analyses
suggest that systematic variation in nuclear DNA content is a more
ubiquitous phenomenon in human cells than was previously appreciated”
(Gillooly, Hein and Damiani 2015, doi:10.1101/cshperspect.a019091).
“Endoreplication [replication of nuclear DNA without cell division] may
be a common and/or functional trait in the somatic cells of humans and
other animals, as it has long been viewed in plants ... Certainly, in
some human cell types (e.g., megakaryocytes), polyploid nuclei are
considered normal and functional. And in other cell types ... (e.g.,
muscle cells), multinucleated cells are common and functional ...
clearly genomes are much more dynamic than was previously thought. We
have moved beyond the view that genomes are immutable blueprints
(Gillooly, Hein and Damiani 2015, doi:10.1101/cshperspect.a019091).
- “The data shown here suggest that perhaps the quantity of nuclear DNA content in human cells is best viewed as a distribution of values that reflects cell size distributions, rather than as a single value. That is, most cells may be of a similar size and contain something close to the characteristic amount of nuclear DNA, but a relatively small number of cells may be large with high amounts of DNA. Changes in the DNA content of somatic cells may occur by endoreplication of individual genes or the entire genome ... [For two cell types examined] the data suggest DNA content has increased as a result of some combination of partial and complete genome duplication” (Gillooly, Hein and Damiani 2015, doi:10.1101/cshperspect.a019091).
- Certain cell types and organs in mammals are characteristically subject to polyploidy. For example: megakaryocytes (cells involved in platelet production in bone marrow) have up to 128 copies of their entire genome; hepatocytes (liver cells constituting some 3/4 of the liver’s mass) typically have 4 to 8 genome copies; trophoblast giant cells in the embryonic outer layer may have up to 1000 copies of the genome; and cardiomyocytes (heart muscle cells) usually have 4 copies of the genome. In humans, about 50% of hepatocytes are polyploid. Some of the proteins involved in the cell cycle appear to play roles in stimulating polyploidy. When polyploid hepatocytes were transplanted into mice whose livers had been partially removed, the new cells regenerated the liver. (See Leslie 2014 for literature references.)
- In one of the liver studies, “when many of the polyploid cells divided, they spawned diploid daughter cells. But often these diploid daughters hadn’t quite returned to normal — many of them had gained or lost an individual chromosome [aneuploidy]. ... Some researchers have proposed that aneuploidy can create useful genetic diversity in a tissue or organ, allowing cells to add a copy of a beneficial gene or throw out a copy of a detrimental one. (See Leslie 2014 for literature references.)
- When the epidermis of an adult fruit fly suffers a puncture wound, a scar forms and surrounding cells go through a process of polyploidization: genomes are duplicated and cells enlarge, without going through cell division. (Other cells fuse, becoming very large with multiple nuclei.) This process continues until there is roughly the same number of genomes as were present in the original, injured tissue — and also the same cell mass. Then the process stops. Something similar occurs in mammals and many other organisms, and is typical of various developmental processes in embryos (Losick, Fox and Spradling 2013). We see in this yet another way the organism proves itself capable of managing its own genome according to context.
- Somatic mitochondrial DNA (mtDNA) mutations are recurrent in specific human tissues. While some tissues in one study had no mtDNA mutations, others had particular mutations in up to 13% of mtDNA copies. Reporting on work by Samuels et al.: “The identified mutations were primarily single-nucleotide variants (SNVs) and were surprisingly recurrent: nine SNVs were found in the same tissue in more than one individual, and the liver and kidney shared almost identical repertoires of SNVs, despite these organs being derived from distinct embryonic tissue types. These recurrent patterns show that the same somatic mtDNA mutations are occurring independently in multiple tissues and individuals” (Burgess 2014).
-
Genetic compensation
“Conventional wisdom holds that modifying a gene to make the encoded protein
inactive — ‘knocking out’ the gene — will have more severe effects than
merely reducing the gene’s expression level. However, there are many cases
in which the opposite occurs. In fact, the knockout of a gene sometimes has
no discernible impact, whereas the reduction of expression (knockdown) of
the same gene causes major defects. [Researchers have now identified] a
molecular mechanism that activates the transcription of genes related to an
inactivated gene, thereby compensating for the knockout”
(Wilkinson 2019, doi:10.1038/d41586-019-00823-5).
“Genetic robustness, or the ability of an organism to maintain fitness in
the presence of harmful mutations, can be achieved via protein feedback
loops. Previous work has suggested that organisms may also respond to
mutations by transcriptional adaptation, a process by which related gene(s)
are upregulated independently of protein feedback loops. However, the
prevalence of transcriptional adaptation and its underlying molecular
mechanisms are unknown. Here, by analysing several models of transcriptional
adaptation in zebrafish and mouse, we uncover a requirement for mutant mRNA
degradation. Alleles that fail to transcribe the mutated gene do not exhibit
transcriptional adaptation, and these alleles give rise to more severe
phenotypes than alleles displaying mutant mRNA decay. Transcriptome
analysis in alleles displaying mutant mRNA decay reveals the upregulation of
a substantial proportion of the genes that exhibit sequence similarity with
the mutated gene's mRNA, suggesting a sequence-dependent mechanism”
(El-Brolosy, Kontarakis, Rossi et al. 2019, doi:10.1038/s41586-019-1064-z).
“The genetic compensation response (GCR) has recently been proposed as a
possible explanation for the phenotypic discrepancies between gene-knockout
and gene-knockdown; however, the underlying molecular mechanism of the GCR
remains uncharacterized. Here, using zebrafish knockdown and knockout models
of the capn3a and nid1a genes, we show that mRNA bearing a
premature termination codon (PTC) promptly triggers a GCR that involves
Upf3a and components of the COMPASS complex. Unlike capn3a-knockdown
embryos, which have small livers, and nid1a-knockdown embryos, which
have short body lengths, capn3a-null and nid1a-null mutants
appear normal. These phenotypic differences have been attributed to the
upregulation of other genes in the same families. By analysing six uniquely
designed transgenes, we demonstrate that the GCR is dependent on both the
presence of a PTC and the nucleotide sequence of the transgene mRNA, which
is homologous to the compensatory endogenous genes. We show that
upf3a (a member of the nonsense-mediated mRNA decay pathway) and
components of the COMPASS complex including wdr5 function in GCR.
Furthermore, we demonstrate that the GCR is accompanied by an enhancement of
histone H3 Lys4 trimethylation (H3K4me3) at the transcription start site
regions of the compensatory genes”
(Ma, Zhu, Shi et al. 2019, doi:10.1038/s41586-019-1057-y).
-
Extracellular genomic DNA fragments
Extracellular vesicles (EVs) are small “containers” budding off from the
cell membrane and carrying a cargo of signaling molecules and other
contents that can subsequently be absorbed into other cells. They have
recently been found to carry fragments of chromosomal and mitochondrial
DNA, and some of these fragments have protein-coding capability. They
presumably also include gene promoters and other regulatory sequences.
- One team of researchers discovered “at least 16434 genomic DNA (gDNA) fragments in the EVs from human plasma”. These fragments showed themselves capable of entering the cytoplasm and nuclei of cells (Cai, Han, Ren et al. 2013).
- The “gDNAs in EVs are transportable between the same or different types of cells, increase the [gDNA-encoded] mRNA and protein expressions in the recipient cells, and have physiological significance to influence function in recipient cells. ... Our present study provides direct evidence that transferred gene[s] can be transcribed in the recipient cells” (Cai, Han, Ren et al. 2013).
- “Secreted DNA may represent a class of signaling molecules that may play an important role in mediating intercellular communication. Moreover, the selective secretion and targeting of DNA among different cells provide a highly regulated complex network under various physiological and pathophysiological conditions” (Cai, Han, Ren et al. 2013).
- See also “Extrachromosomal DNA” under PRE-TRANSCRIPTIONAL DECISION-MAKING above.
-
Extracellular mRNA and miRNA
“The exosome-mediated transfer of genetic information in the form of
microRNAs (miRNAs) or mRNAs has been primarily shown in vitro; a new
study now suggests that such transfer occurs between the haematopoietic
system and the brain in vivo”. Transfer of the RNAs also occurred
into the liver, lung, and small intestine. The transfer occurs in
blood-borne vesicles, but the exact means of entry into other tissues
remains unclear (Burgess 2014, reporting on research by K. Ridder et al.
2014).
“RNA, once thought to exist only in cells, is now known to travel to tissues
all over the body through the blood, under the protection of tiny lipid sacs
known as extracellular vesicles. The study of this extracellular RNA (exRNA)
has led to a quiet revolution in biology, as scientists endeavour to
understand why cells release RNA, and how the molecules might be used to
improve the detection and treatment of disease ... One provocative study
that linked RNA in what we eat to gene expression has kick-started vigorous
efforts to learn the language in which our food speaks to us. The link,
however, remains unproven. Related efforts are focusing on how RNA in breast
milk affects infant health” (Brody 2020, doi:10.1038/d41586-020-01762-2).
-
Mitochondria
Mitochondria present a whole additional world of genes, gene expression, translation, and so on. This is barely touched on in this document. But here are a few notes hinting at the significant phenomena thus overlooked:
“Mitochondria are essential organelles that act as energy conversion
powerhouses and metabolic hubs. Their gene expression machineries combine
traits inherited from prokaryote ancestors and specific features acquired
during eukaryote evolution. Mitochondrial research has wide implications
ranging from human health to agronomy. We highlight recent advances in
mitochondrial translation. Functional, biochemical, and structural data have
revealed an unexpected diversity of mitochondrial translation systems,
particularly of their key players, the mitochondrial ribosomes
(mitoribosomes)” (Waltz and Giegé 2020, doi:10.1016/j.tibs.2019.10.004).
“Metazoan cells possess two genomes (i.e., nuclear and mitochondrial), but
the nuclear DNA was thought to exclusively encode regulators of both
genomes. Recently, Bérénice A. Benayoun and Changhan Lee found that a small
peptide encoded in the mitochondrial genome, MOTS‐c, directly regulates the
nuclear genome, revealing an integrated bi‐genomic basis of gene
regulation” (Table of contents entry for Benayoun and Lee 2019,
doi:10.1002/bies.201900046).
“MOTS‐c (mitochondrial open reading frame of the 12S ribosomal RNA type‐c)
is a recently identified peptide encoded within the mitochondrial 12S
ribosomal RNA gene that has metabolic functions. Notably, MOTS‐c can
translocate to the nucleus upon metabolic stress (e.g., glucose restriction
and oxidative stress) and directly regulate adaptive nuclear gene expression
to promote cellular homeostasis. It is hypothesized that cellular fitness
requires the coevolved mitonuclear genomes to coordinate adaptive responses
using gene‐encoded factors that cross‐regulate the opposite genome. This
suggests that cellular gene expression requires the bipartite split genomes
to operate as a unified system, rather than the nucleus being the sole
master regulator” (Benayoun and Lee 2019, doi:10.1002/bies.201900046).
-
Physiology of the cell
Transcription factors are the classic regulators of transcription. Because
they are themselves proteins resulting from the expression of genes, the
transcription factor activity can (if we turn a blind eye to much of the
contents of this document) be seen as a fairly direct way in which the
genome regulates its own expression. Widely ignored is the role of cellular
physiology in influencing gene expression. But that seems to be changing.
- “Organisms must be able to rapidly alter gene expression in response to changes in their nutrient environment. This review summarizes evidence that epigenetic modifications of chromatin depend on particular metabolites of intermediary metabolism, enabling the facile regulation of gene expression in tune with metabolic state. Nutritional or dietary control of chromatin is an often-overlooked, yet fundamental regulatory mechanism directly linked to human physiology. Nutrient-sensitive epigenetic marks are dynamic, suggesting rapid turnover” (Huang, Cai and Tu 2015, doi:10.1016/j.ceb.2015.05.004).
- “Metabolites may also influence the recruitment of transcriptional regulatory complexes to DNA ... Histone modifiers are also themselves subject to metabolite-sensitive modifications” (Huang, Cai and Tu 2015, doi:10.1016/j.ceb.2015.05.004).
- One research group analyzed the regulation of carbon metabolism in the bacterium, E. coli. “Our results show that the transcriptional response of the network is controlled by the physiological state of the cell and the signaling metabolite cyclic AMP. The absence of a strong regulatory effect of transcription factors suggests that they are not the main coordinators of gene expression changes during growth transitions, but rather that they complement the effect of global physiological control mechanisms” (Berthoumieux, de Jong, Baptist et al. 2013, doi8:10.1038/msb.2012.70). In describing the research, the editors of Science wrote: “it was not the binding of transcription factors to particular target genes that produced the changes in gene expression in the bacteria, but instead global changes in transcription and translation mediated by changes, for example, in the abundance of RNA polymerase, ribosomes, and the pools of available amino acids and nucleotides. Mathematical modeling used to measure the relative contributions of specific transcriptional control and global changes in physiological state showed the primary mechanism to be the latter, which ironically is almost never accounted for in diagrams of cellular regulatory networks” (note in “Editors’ Choice”, March 15, 2013).
-
Cell-to-cell variability
Researchers have only recently (I am writing in 2016) been able to assay
various gene-related processes at the level of the individual cell. One of
the surprises has been the great variability even among cells of the “same”
type within the same tissue. This, of course, greatly increases the
complexity of numerous issues relevant to gene regulation.
-
Martinez-Jimenez and Odom (2016) list a number of factors, both intrinsic
to the individual cell and extrinsic, that are related to the “random”
aspects of gene expression:
- Intrinsic factors: Transcriptional functions, or bursts (stochasticity); chromatin architecture (chromosomal conformation and epigenetic marks); and mRNA processing (transport, translation and degradation rates).
- Extrinsic factors: Cell size and ploidy (number of sets of chromosomes in a cell); cellular reprogramming and specific-cell-type networks; microenvironment (including pathogen responses); cell cycle (symmetric and asymmetric cell division); basal machinery availability (for example, basic transcriptional and translational factors).
-
Martinez-Jimenez and Odom (2016) list a number of factors, both intrinsic
to the individual cell and extrinsic, that are related to the “random”
aspects of gene expression:
-
Symbiosis
Not all important gene expression is “our own”. Our bodies depend on huge
numbers of microorganisms, whose lives are more or less fully integrated
into ours. “The collective genetic potential (metagenome) of the human
microbiome is orders of magnitude more than the human genome, and it
profoundly affects human health and disease in ways we are only beginning
to understand” (doi:10.1016/j.tig.2012.09.005).
-
Microbiome
Remarkably, “one-third of the metabolites in the blood are coming from
gut bacteria”, according to Phillip Hylemon, a microbiologist and
immunologist at Virginia Commonwealth University in Richmond (quoted in
Bourzac 2014). Altogether, our microorganisms “function as another
organ, complementing and interacting with human metabolism in ways not
fully understood” (Gravitz 2012).
“The human microbiome encodes a second genome that dwarfs the genetic
capacity of the host. Microbiota-derived small molecules can directly
target human cells and their receptors or indirectly modulate host
responses through functional interactions with other microbes in their
ecological niche. Their biochemical complexity has profound implications
for nutrition, immune system development, disease progression, and drug
metabolism, as well as the variation in these processes that exists
between individuals”
(Shine and Crawford 2021, doi:10.1146/annurev-biochem-080320-115307)
“Maintaining intestinal homeostasis is a key prerequisite for a healthy
gut. Recent evidence points out that microRNAs (miRNAs) act at the
epicenter of the signaling networks regulating this process. The fine
balance in the interaction between gut microbiota, intestinal epithelial
cells, and the host immune system is achieved by constant transmission of
signals and their precise regulation. Gut microbes extensively
communicate with the host immune system and modulate host gene
expression. On the other hand, sensing of gut microbiota by the immune
cells provides appropriate tolerant responses that facilitate the
symbiotic relationships. While the role of many regulatory proteins,
receptors and their signaling pathways in the regulation of the
intestinal homeostasis is well documented, the involvement of non-coding
RNA molecules in this process has just emerged”
(Belcheva 2017, doi:10.1002/bies.201600200).
- The gene expression in our intestinal flora plays an important role in our lives. For example, Japanese individuals can digest seaweed carbohydrates more easily than North Americans. This appears to be due to a horizontal gene transfer among microbes, giving the Japanese intestinal microbes the ability to produce two enzymes important for digesting seaweed carbohydrates. The research community will have to “come to terms with the extent to which human genetic effects on diet might be overwhelmed by the bacteria we carry ... ‘It’s something I use in my slide presentations now to worry geneticists’” (Eisenstein 2010, quoting Jeremy Nicholson, a biological chemist at Imperial College London).
- Moreover, microorganisms can directly play into the host organism’s genetic performances. When a mouse is suffering a listeria infection, its gut microbes alter the expression of the mouse’s microRNAs in the intestinal ileum. That is, infected mice possessing normal intestinal flora express their own microRNAs differently than do infected mice lacking a normal microbiome (Archambaud et al. 2013). This, of course, has implications for all the gene expression influenced in turn by the regulatory activity of those microRNAs.
-
The December 2013 issue of Cell had the rather surprising title,
“Toward Effective Probiotics for Autism and Other
Neurodevelopmental Disorders” (Gilbert et al. 2013, reporting on
work by Hsiao et al. 2013). The authors of the original research
worked with mice in which an autism-like condition had been
induced. The mice had “reduced intestinal integrity through altered
gut bacterial community”, and the bacterial metabolites varied
considerably from normal mice. Moreover, when the mice “were fed
the probiotic, Bacteroides fragilis, a gut microbe with positive
effects on the immune system, the abundance of 34% of these
metabolites changed back, gut barrier integrity was improved, the
gut-microbiome was restored to a state similar to control mice, and
a number of ASD [autism-spectrum disorder]-related behavioral
abnormalities were ameliorated”.
Perhaps rather ambitiously, Gilbert and co-authors write: “Several reports indicate that probiotics can treat anxiety and posttraumatic stress disorder in mouse models ... Therapies that target our microbial side may hold the key to making progress against a wide range of notoriously difficult psychiatric illnesses”.
- “Gut microbes and the fatty acids they produce can regulate gene expression by influencing the 3D shape of their hosts’ DNA ... Gut microbes mediate chemical changes to histone proteins, which in turn regulate gene expression by binding to DNA and altering its 3D conformation” (Nature editors 2016, doi:10.1038/540011e).
- “We show here in C. elegans that nitric oxide derived from resident bacteria promotes widespread S-nitrosylation of the host proteome. We further show that microbiota-dependent S-nitrosylation of C. elegans Argonaute protein (ALG-1) — at a site conserved and S-nitrosylated in mammalian Argonaute 2 (AGO2) — alters its function in controlling gene expression via microRNAs. By selectively eliminating nitric oxide generation by the microbiota or S-nitrosylation in ALG-1, we reveal unforeseen effects on host development. Thus, the microbiota can shape the post-translational landscape of the host proteome to regulate microRNA activity, gene expression, and host development” (Seth, Hsieh, Jamal et al. 2019, doi:10.1016/j.cell.2019.01.037).
-
Viruses
- “In the absence of bacteria, mammalian intestinal viruses promote gut homeostasis and protect the intestine from injury and from pathogenic bacteria” (Wang and Pfeiffer 2014, citing work by Kernbauer et al. 2014).
-
Microbiome
[This section is currently trivial compared to what it could be. It probably never will be more fully developed, because in fact the story of integrated cellular activity in gene regulation is the story narrated by virtually everything in this document (together with much that is not touched on here).]
A rapidly growing literature focuses not on separate processes of regulation,
but rather on their integration. “Transcription, translation and degradation
are often extensively coupled and may frequently regulate each other through
feedback loops” (Vogel and Marcotte 2012). “Until recently the process of gene
expression was predominantly considered to be a series of isolated steps, each
controlled by separate regulatory mechanisms and networks. However, it is
becoming increasingly clear that the different phases of the process are
coupled” (Dahan, Gingold and Pilpel 2011). For example:
“We are beginning to get glimpses of the
fact that epigenetic mechanisms are far more complex and
likely to involve the interplay of the genome, physiological
state of the organism, nervous system, and other environmental
input” (Pirrotta 2016, doi:10.1101/cshperspect.a019547).
More and more, reports on events pertaining to gene expression read rather like
this abstract: “Vascular Endothelial Growth Factor A (VEGF-A) is a potent
secreted mitogen crucial for physiological and pathological angiogenesis.
Post-transcriptional regulation of VEGF-A occurs at multiple levels. Firstly,
alternative splicing gives rise to different transcript variants encoding
diverse isoforms that exhibit distinct biological properties with regard to
receptor binding and extra-cellular localization. Secondly, VEGF-A mRNA
stability is regulated by effectors such as hypoxia or growth factors through
the binding of stabilizing and destabilizing proteins at AU-rich elements
located in the 3' untranslated region. Thirdly, translation of VEGF-A mRNA is a
controlled process involving alternative initiation codons, internal ribosome
entry sites, an upstream open reading frame, miRNA targeting and a riboswitch
in the 3' untranslated region. These different levels of regulation cooperate
for the crucial fine-tuning of the expression of VEGF-A variants” (Arcondéguy,
Lacazette, Millevoi et al. 2013). Why should we speak about this solely as
“regulation of gene expression”? It can be seen from that standpoint, but
equally well from many other standpoints. In reality, it’s a picture of
diverse, coordinated cellular activities relating to the needs of the organism
— a picture in which DNA sequences find their place alongside all the other
things going on.
“Translation factors have traditionally been viewed as proteins that drive
ribosome function and ensure accurate mRNA translation. Recent discoveries have
highlighted that these factors can also moonlight in gene regulation, but
through functions distinct from their canonical roles in protein synthesis.
Notably, the additional functions that translation factors encode are diverse,
ranging from transcriptional control and extracellular signaling to RNA
binding, and are highly regulated in response to external cues and the
intrinsic cellular state. Thus, this multifunctionality of translation factors
provides an additional mechanism for exquisite control of gene expression”
(Farache, Antine and Lee 2022, doi:10.1016/j.tcb.2022.03.006).
-
“We use [the miRNA] LIN-28 as an example to illustrate how distinct
regulatory systems, including miRNAs and multiple protein stability
mechanisms, work at different levels to target expression of a given gene
and provide tissue-specific and stage-specific regulation of gene
expression”. In particular:
“miRNA pathways converge with diverse non-miRNA regulatory mechanisms to regulate common targets.
“Convergent regulation at different levels of gene expression imparts tissue-specific functions, synergy, and precise temporal gene regulation.
“Proteolytic pathways provide a strong commitment in gene regulation since proteolysis is irreversible without new protein synthesis.
“Genetic redundancy remains a critical barrier in understanding genomic architecture and regulatory networks.
“Genes with known functions may have additional important non-canonical functions” (Weaver and Han 2017, doi:10.1016/j.tig.2017.09.009).
This illustrates a good deal about the principle of wholeness in organisms — the way processes are globally interwoven in the interests of the overall unity of the organism. For example, any given function of a molecule tends to involve roles by many other sorts of molecule; and any given molecule tends to have multiple functions. To imagine how this works is to imagine all activity under the “guiding hand” of the whole.
-
“Coupling between transcription and downstream RNA-processing events is
predominantly achieved by the recruitment of different processing factors
to the nascent RNA during transcription, and this takes place via an
interaction with RNA polymerase II (RNAPII). RNAPII can adopt different
phosphorylation states during transcription, forming a pattern that
determines the recruitment of alternative RNA-processing molecules. In this
respect the polymerase serves as dynamic docking platform that orchestrates
different RNA-processing events during transcription. An interesting
example is the connection between transcription and splicing. The
phosphorylation status of RNAPII appears to be related to the rate of
transcription, which in turn affects (and is affected by) splicing events
along transcripts” (Dahan, Gingold and Pilpel 2011). The authors discuss
the following couplings in particular:
- Transcription and RNA localization
- Transcription and translation
- Transcription and mRNA degradation
- Translation and mRNA degradation
-
“The KZFP/KAP1 (KRAB zinc finger proteins/KRAB-associated protein 1) system
plays a central role in repressing transposable elements and maintaining
parent-of-origin DNA methylation at imprinting control regions during the
wave of genome-wide reprogramming that precedes implantation. In naïve
murine embryonic stem cells, the genome is maintained highly hypomethylated
by a combination of TET-mediated active demethylation and lack of de novo
methylation, yet KAP1 is tethered by sequence-specific KZFPs to ICRs and TEs
where it recruits histone and DNA methyltransferases to impose
heterochromatin formation and DNA methylation.
“Our results indicate that the KZFP/KAP1 complex maintains heterochromatin and DNA methylation at ICRs [imprinting control regions] and TEs [transposable elements] in naïve embryonic stem cells partly by protecting these loci from TET-mediated demethylation. Our study further unveils an unsuspected level of complexity in the transcriptional control of the endovirome by demonstrating often integrant-specific differential influences of histone-based heterochromatin modifications, DNA methylation and 5mC oxidation in regulating TEs expression” (Coluccio, Ecco, Duc 2018, doi:10.1186/s13072-018-0177-1).
-
Example: Coupling of transcription and mRNA degradation
Many transcription factors regulate many genes, and those genes may be
post-transcriptionally regulated (degraded) by miRNAs. A transcription
factor and an miRNA often regulate a common set of targets. More than
that, the transcription factor regulates the miRNA, and the miRNA regulates
the transcription factor. And this situation can be multiplied in
complexity, as multiple transcription factors, miRNAs, and other molecules
become involved in the various mutual interactions — something that is
true, for example, in the regulation of circadian rhythms.
-
Example: Relation between transcription factor binding, chromatin
modifications, and DNA methylation
“Transcription factors (TFs) and epigenetic modifications play crucial roles
in the regulation of gene expression, and correlations between the two types
of factors have been discovered ... We implemented statistical analyses to
illustrate that epigenetic modifications are predictive of TF binding
affinities, without the need of [DNA] sequence information. Intriguingly, by
considering genome locations relative to transcription start sites (TSSs) or
enhancer midpoints, our analyses show that different locations display
various relationship patterns. For instance, [the histone modifications]
H3K4me3, H3k9ac and H3k27ac contribute more in the regions near TSSs,
whereas H3K4me1 and H3k79me2 dominate in the regions far from TSSs. DNA
methylation plays relatively important roles when close to TSSs than in
other regions. In addition, the results show that epigenetic modification
models for the predictions of TF binding affinities are cell line-specific.
Taken together, our study elucidates highly coordinated, but location- and
cell type-specific relationships between epigenetic modifications and
binding affinities of TFs” (Liu, Jin and Zhou 2015, doi:10.1093/nar/gkv255).
-
Example: Some factors involved in heart development
“Heart development is exquisitely sensitive to the precise temporal
regulation of thousands of genes that govern developmental decisions during
differentiation...Here, we interrogated the transcriptome and several
histone modifications across the genome during defined stages of cardiac
differentiation. We find distinct chromatin patterns that are coordinated
with stage-specific expression of functionally related genes, including
many human disease-associated genes. Moreover, we discover a novel
preactivation chromatin pattern at the promoters of genes associated with
heart development and cardiac function. We further identify stage-specific
distal enhancer elements and find enriched DNA binding motifs within these
regions that predict sets of transcription factors that orchestrate cardiac
differentiation” (Wamstad, Alexander, Truty et al. 2012).
- Example: Antisense transcription, RNA splicing, noncoding RNA, and intronic promoter “[Our] results indicate that [transcription factor] LEF1 gene expression is attenuated by an antisense non-coding RNA and that this NAT [natural antisense transcript] function is regulated by the balance between its spliced and unspliced forms”. The details: “we have analyzed the role of antisense transcription in the control of LEF1 transcription factor expression. A NAT is transcribed from a promoter present in the first intron of LEF1 gene and undergoes splicing in mesenchymal cells. Although this locus is silent in epithelial cells, and neither NAT transcript nor LEF1 mRNA are expressed, in cell lines with an intermediate epithelial-mesenchymal phenotype presenting low LEF1 expression, the NAT is synthesized and remains unprocessed. Contrarily to the spliced NAT, this unspliced NAT down-regulates the main LEF1 promoter activity and attenuates LEF1 mRNA transcription. Unspliced LEF1 NAT interacts with LEF1 promoter and facilitates PRC2 binding to the LEF1 promoter and trimethylation of lysine 27 in histone 3. Expression of the spliced form of LEF1 NAT in trans prevents the action of unspliced NAT by competing for interaction with the promoter” (Beltran, Aparicio-Prat, Mazzolini et al. 2015, doi:10.1093/nar/gkv502).
-
Example: Vascular endothelial growth factor
“Vascular Endothelial Growth Factor A (VEGF-A) is a potent secreted mitogen
[substance tending to trigger cell division and mitosis] crucial for
physiological and pathological angiogenesis [formation of blood vessels].
Post-transcriptional regulation of VEGF-A occurs at multiple levels.
Firstly, alternative splicing gives rise to different transcript variants
encoding diverse isoforms that exhibit distinct biological properties with
regard to receptor binding and extra-cellular localization. Secondly,
VEGF-A mRNA stability is regulated by effectors such as hypoxia or growth
factors through the binding of stabilizing and destabilizing proteins at
AU-rich elements located in the 3' untranslated region. Thirdly, translation
of VEGF-A mRNA is a controlled process involving alternative initiation
codons, internal ribosome entry sites (IRESs), an upstream open reading
frame (uORF), miRNA targeting and a riboswitch in the 3' untranslated
region. These different levels of regulation cooperate for the crucial
fine-tuning of the expression of VEGF-A variants” (Arcondéguy, Lacazette,
Millevoi et al. 2013, doi:10.1093/nar/gkt539).
There’s also a role for noncoding RNA: “It was recently shown that a non-coding regulatory RNA, mapped in the 5' UTR of VEGF-A mRNA, plays a function in tumour development by affecting the expression of other genes independently of VEGF-A translation”.
And a G-quadruplex structure located within a VEGF-A internal ribosome entry site (IRES-A) appears to play a role as well. Mutations disrupting the structure “inactivated IRES-A function, suggesting the requirement of this structure to maintain IRES-A activity ... IRESs ensure translation activation of mRNA during stress”.
-
Example: Nuclear receptor, structural protein, transcription factors,
histone variant, and nucleosome positioning
The integration of many contextual factors in regulation of gene expression
helps to explain how it is that any given factor commonly can play a role in
“opposite” regulatory functions — that is, in both activation and repression
of a gene. Thus, the progesterone receptor, which (when bound by its
hormone ligand) “normally” activates its target genes, can also repress them
by recruiting BRG1, which is part of the SWI/SNF chromatin remodeling
complex, and the HP1γ-LSD1 complex (another remodeler of chromatin
structure). “In contrast to what is observed during gene activation, only
BRG1 and not the BAF complex is recruited to repressed promoters, likely due
to local enrichment of the pioneer [transcription] factor FOXA1. BRG1
participates in gene repression by interacting with [histone variant] H1.2,
facilitating its deposition and stabilizing nucleosome positioning around
the transcription start site”
(Nacht, Pohl, Zaurin et al. 2016, doi:10.15252/embj.201593260).
-
Example: Interactions among neighboring genes, promoters, enhancers,
splice sites, long noncoding RNAs, and transcription
“Mammalian genomes are pervasively transcribed to produce thousands of long
non-coding RNAs (lncRNAs) ... lncRNA expression is often correlated with the
expression of nearby genes ... some gene promoters have been proposed to
have dual functions as enhancers, and the process of transcription itself
may contribute to gene regulation by recruiting activating factors or
remodelling nucleosomes. Here we use genetic manipulation in mouse cell
lines to dissect 12 genomic loci that produce lncRNAs and find that 5 of
these loci influence the expression of a neighbouring gene in cis.
Notably, none of these effects requires the specific lncRNA transcripts
themselves and instead involves general processes associated with their
production, including enhancer-like activity of gene promoters, the process
of transcription, and the splicing of the transcript. Furthermore, such
effects are not limited to lncRNA loci: we find that four out of six
protein-coding loci also influence the expression of a neighbour. These
results demonstrate that cross-talk among neighbouring genes is a prevalent
phenomenon that can involve multiple mechanisms and cis-regulatory signals,
including a role for RNA splice sites”
(Engreitz, Haines, Perez et al. 2016, doi:10.1038/nature20149).
-
Example: Aspects of chromatin organization
“During differentiation of embryonic stem cells, chromatin reorganizes to
establish cell type-specific expression programs. Here, we have dissected
the linkages between DNA methylation (5mC), hydroxymethylation (5hmC),
nucleosome repositioning, and binding of the transcription factor CTCF
during this process. ... we found that the interplay between these factors
depends on their genomic context. The mostly unmethylated CpG islands have
reduced nucleosome occupancy and are enriched in cell type-independent
binding sites for CTCF. The few remaining methylated CpG dinucleotides are
preferentially associated with nucleosomes. In contrast, outside of CpG
islands most CpGs are methylated, and the average methylation density
oscillates so that it is highest in the linker region between nucleosomes.
Outside CpG islands, binding of TET1, an enzyme that converts 5mC to 5hmC,
is associated with labile ... nucleosomes. Such nucleosomes are poised for
eviction in embryonic stem cells and become stably bound in differentiated
cells where the TET1 and 5hmC levels go down. This process regulates a
class of CTCF binding sites outside CpG islands that are occupied by CTCF
in embryonic stem cells but lose the protein during differentiation” (Teif,
Beshnova, Vainshtein et al. 2014).
-
Example: long noncoding RNA, R-loops, DNA demethylation, antisense
transcription
R-loops are DNA–RNA hybrids enriched at CpG islands (CGIs) that can regulate
chromatin states ... Here we show that GADD45A (growth arrest and DNA damage
protein 45A) binds directly to R-loops and mediates local DNA demethylation
by recruiting TET1 (ten-eleven translocation 1). Studying the tumor
suppressor TCF21, we find that antisense long noncoding TARID (TCF21
antisense RNA inducing promoter demethylation) forms an R-loop at the TCF21
promoter. Binding of GADD45A to the R-loop triggers local DNA demethylation
and TCF21 expression. TARID transcription, R-loop formation, DNA
demethylation, and TCF21 expression proceed sequentially during the cell
cycle ... Genomic profiling in embryonic stem cells identifies thousands of
R-loop-dependent TET1 binding sites at CGIs. We propose that GADD45A is an
epigenetic R-loop reader that recruits the demethylation machinery to
promoter CGIs.
(Arab, Karaulanov, Musheev et al. 2019; doi:10.1038/s41588-018-0306-6)
-
RNA splicing and RNA editing
“Here, we systematically test the influence of splice rates on RNA-editing
using reporter genes but also endogenous substrates. We demonstrate for the
first time that the extent of editing is controlled by splicing kinetics
when editing is guided by intronic elements. In contrast, editing sites that
are exclusively defined by exonic structures are almost unaffected by the
splicing efficiency of nearby introns. In addition, we show that editing
levels in pre- and mature mRNAs do not match. This phenomenon can in part be
explained by the editing state of an RNA influencing its splicing rate but
also by the binding of the editing enzyme ADAR that interferes with
splicing” (Licht, Kapoor, Mayrhofer and Jantsch 2016, 10.1093/nar/gkw325).
-
DNA replication and transcription
- “We find it curious that replication timing has been left out of important discussions of epigenetics. ... Replication timing is a mitotically stable yet cell-type specific feature of chromosomes. Chromatin is assembled at the replication fork and different types of chromatin are assembled at different times during S-phase. Every multi-cellular organism studied to date exhibits a strong positive correlation between early replication and transcription. At the same time, changes in replication timing are not directly influenced by nor do they have a direct influence on transcription but rather define a level of higher-order organization of the genome, which is thought to affect transcriptional competence independent of transcription per se. Replication timing is therefore more in line with the concept of epigenetic inheritance than most histone modifications: indeed, replication defines mitotic inheritance. In fact, the time point of commitment for X chromosome inactivation in mammals is independent of transcriptional down-regulation but is coincident with a nearly chromosome-wide change in replication timing of the inactive X, which is one of the best-conserved characteristics of mammalian X chromosome inactivation. Recent work demonstrates convincingly that segments of all autosomes undergo similar changes in replication timing during cell fate determination. Hence, changes in replication-timing profiles reveal chromosome segments that undergo large changes in organization during differentiation and may provide a handle into previously impenetrable levels of chromosome organization and their relationship to cellular identity” (Hiratani, Ichiro and David M. Gilbert (2009).
-
Transcription factors, co-factors, and enhancers
- Researchers fused the DNA-binding domains of 812 Drosophila transcription factors (TFs) and co-factors to the GAL4 gene, and then recruited the various fusions to 24 enhancer contexts in order to measure the resulting enhancer activities. “Most factors were functional in at least one context, yet their contributions differed between contexts and varied from repression to activation (up to 289-fold) for individual factors. Based on functional similarities across contexts, we define 15 groups of TFs that differ in developmental functions and protein sequence features. Similar TFs can substitute for each other, enabling enhancer re-engineering by exchanging TF motifs, and TF–cofactor pairs cooperate during enhancer control and interact physically. Overall, we show that activators and repressors can have diverse regulatory functions that typically depend on the enhancer context” (Stampfel, Kazmar, Frank et al. 2015, doi:10.1038/nature15545).
-
Signaling pathways
- “It is becoming increasingly obvious that cellular signaling pathways control gene expression programs at multiple levels, from transcription through RNA processing and finally protein production. ... First, at the transcriptional level, the response to a particular pathway can be highly coordinated, with pols [polymerases] I, II, and III being controlled simultaneously via a diverse range of mechanisms. Transcription factors ... serve to integrate these responses to the signaling pathways. Second, in addition to RNA production, RNA processing and translation are key steps that are controlled by the same pathways. Part of this control is exerted through the transcriptional induction of ribosomal proteins and rRNAs but also there are direct effects of the signaling kinases in controlling the activities of components of the RNA processing, RNA splicing and protein translation machinery. Third, the establishment of gene expression programs in response to signaling is complex ...” (White and Sharrocks 2010).
-
Stem cells
- A research team reviewing the role of transcriptional activators and other factors in pluripotent and stem cells writes: “The emerging picture reveals an elaborate mechanism of gene regulation in which no single protein or protein complex is more important than the rest; instead, they appear to be physically and functionally intertwined” (Fong, Cattoglio, Yamaguchi and Tjian 2012).
- “Adult stem cells are important for mammalian tissues, where they act as a cell reserve that supports normal tissue turnover and can mount a regenerative response following acute injuries. Quiescent stem cells are well established in certain tissues, such as skeletal muscle, brain, and bone marrow. The quiescent state is actively controlled and is essential for long-term maintenance of stem cell pools ... Recent studies revealed that quiescent stem cells have a discordance between RNA and protein levels, indicating the importance of post-transcriptional mechanisms, such as alternative polyadenylation, alternative splicing, and translation repression, in the control of stem cell quiescence.
-
Chromatin structure
- “DNA methylation, histone variants and modifications, and nucleosome positioning work together and generate active or inactive chromatin configurations” (Kelly, Miranda, Liang, Berman et al. (2010).
-
The ribonome
The “ribonome” is the entire collection of RNA molecules in a cell or
organism, along with the diverse proteins that associate with them.
- Australian researcher John Mattick argues that, taken together, RNAs constitute the true “computational engine of the cell” (Mattick 2009; Mattick et al. 2009). This “engine” includes numerous small RNAs whose functions are the result, not simply of their transcription from DNA, but of their elaborate processing and restructuring within nucleus and cytoplasm. RNA in general “is known or strongly implicated to be involved in the regulation of gene expression (both protein-coding and noncoding) at all levels in animals, creating extraordinarily complex hierarchies of interacting controls. This includes chromatin modification and associated epigenetic memory, transcription, alternative splicing, RNA modification, RNA editing, mRNA translation, RNA stability, and cellular signal transduction and trafficking pathways." (Mattick 2007)
-
Membrane architecture of the cell
“Cellular organization in general and membrane-mediated
compartmentalization in particular are constitutive of the biological
‘meaning’ of any newly synthesized protein (and thus gene), which is either
properly targeted within the context of cellular compartmentalization or
quickly condemned to rapid destruction (or cellular ‘mischief’). At the
level of the empirical materiality of real cells, genes ‘show up’ as
indeterminate resources...If cellular organization is ever lost, neither
‘all the king’s horses and all the king’s men’ nor any amount of DNA
could put it back together again” (Moss 2003).
- “An increasing corpus of research has demonstrated that membrane shape, generated either by the external environment of the cell or by intrinsic mechanisms such as cytokinesis and vesicle or organelle formation, is an important parameter in the control of diverse cellular processes ... Membrane curvature – far from being a passive consequence of the physical environment and the internal protein activity of a cell – is an important signal that controls protein affinity and enzymatic activity to ensure robust forward progression of key processes within the cell” (Cail and Drubin 2023, doi:10.1016/j.tcb.2022.09.004).
[This section scarcely begun.]
Unity, complexity, interactivity, holism
“Our initial ‘modular’ notion of a gene has been challenged by the realization
that: (i) multiple layers of regulatory information permeate the transcribed
region; (ii) eukaryotic genomes are pervasively transcribed, generating an
ensemble of transcripts from any given locus; (iii) each of these transcripts
might in turn undergo multiple rounds of cleavage to generate even greater
complexity; and (iv) this panoply of transcripts can perform diverse biological
roles. The overlapping nature of the genetic information and transcripts
associated with a single locus limits the value of studies of any component in
isolation. We therefore suggest that each gene must now be regarded as a
system, comprising a genomic region with the corresponding network of control
regions and ensemble of transcripts" (Tuck and Tollervey 2011).
And that is not to mention about 98 percent of the regulatory goings-on briefly pointed to in all the foregoing — goings-on that bring virtually every aspect of the organism to bear upon DNA and its transcripts.
Variability, change, dynamism
Re: Changes in yeast gene transcripts during development: “Our analyses reveal
extensive changes to both the coding and noncoding transcriptome, including
altered 5' ends, 3' ends, and splice sites. Additionally, 3910 (46.5%)
unannotated [previously undocumented] expressed segments were identified.
Interestingly, subsets of unannotated RNAs are located across from introns
(anti-introns) or across from the junction between two genes (anti-intergenic
junctions). Many of these unannotated RNAs are abundant and exhibit
sporulation-specific changes in expression patterns. ... Our high-resolution
transcriptome analyses reveal that coding and noncoding transcript
architectures are exceptionally dynamic in S. cerevisiae and suggest a
vast array of novel transcriptional and post-transcriptional control mechanisms
that are activated upon meiosis and sporulation”. “Functionally distinct
changes in [transcript] architecture frequently occur in response to signaling
cascades. Therefore, identification of dynamic changes to transcript
architecture often requires the study of a dynamic process” (Guisbert, Zhang,
Flatow et al. 2012).
In sum: the authors speak of “dramatic architecture changes” and an “unexpectedly dynamic” transcriptome. “Not only do unannotated transcripts exhibit dynamic expression patterns in terms of both architecture and overall expression, but the regulation of this expression appears to play a critically important role in the progression of meiosis”. “We predict that many other stress conditions or developmental cascades will also induce novel examples of transcriptome regulation”.
The organism is an activity, not a collection of things
This truth is emerging on all fronts. An illustration: researchers looked at a
“spectacular example of convergent evolution and phenotypic plasticity” —
namely the independent arising of queen and worker castes in bees, ants, and
wasps. The common notion that such cases must involve deeply conserved genes
was not supported by this work. Rather, “Overall, we found few shared caste
differentially expressed transcripts across the three social lineages. However,
there is substantially more overlap at the levels of pathways and biological
functions. Thus, there are shared elements but not on the level of specific
genes. Instead, the toolkit appears to be relatively “loose,” that is,
different lineages show convergent molecular evolution involving similar
metabolic pathways and molecular functions but not the exact same genes.
Additionally, our paper wasp data do not support a complementary hypothesis
that “novel” taxonomically restricted genes are related to caste differences”
(Berens, Hunt and Toth 2015, doi:10.1093/molbev/msu330).
Genes, in other words, are not master controllers or bearers of controlling instructions, but rather represent resources that evolving organisms can employ in their own ways. The same genes can be put to very different uses, and different genes can be caught up in the service of similar ends. The determination of a gene’s meaning is made by the organism as a whole, based on its patterns of activity.
This document: https://bwo.life/org/support/genereg.htm
Steve Talbott :: How the Organism Decides What to Make of Its Genes